
Abstract
Adult mesenchymal stem cells are a promising source for cell therapies and tissue engineering applications. Current procedures for banking of human bone-marrow mesenchymal stem cells (hBM-MSCs) require cell isolation and expansion, and thus the use of large amounts of animal sera. However, animal-derived culture supplements have the potential to trigger infections and severe immune reactions. The aim of this study was to investigate an optimized method for cryopreservation of human bone-marrow fragments for application in cell banking procedures where stem-cell expansion and use are not immediately needed. Whole trabecular fragments enclosing the bone marrow were stored in liquid nitrogen for 1 year in a cryoprotective solution containing a low concentration of dimethyl sulfoxide and a high concentration of human serum (HuS). After thawing, the isolation, colony-forming-unit ability, proliferation, morphology, stemness-related marker expression, cell senescence, apoptosis, and multi-lineage differentiation potential of hBM-MSCs were tested in media containing HuS compared with hBM-MSCs isolated from fresh fragments. Human BM-MSCs isolated from cryopreserved fragments expressed MSC markers until later passages, had a good proliferation rate, and exhibited the capacity to differentiate toward osteogenic, adipogenic, and myogenic lineages similar to hBM-MSCs isolated from fresh fragments. Moreover, the cryopreservation method did not induce cell senescence or cell death. These results imply that minimal processing may be adequate for the banking of tissue samples with no requirement for the immediate isolation and use of hBM-MSCs, thus limiting cost and the risk of contamination, and facilitating banking for clinical use. Furthermore, the use of HuS for cryopreservation and expansion/differentiation has the potential for clinical application in compliance with good manufacturing practice standards.
Introduction
S
Among different tissue sources for MSCs, bone-marrow-derived MSCs (BM-MSCs) represent a suitable tool for the treatment of various diseases. A major challenge is to develop a cryopreservation protocol that allows long-term storage without affecting the characteristics of the cells, thus encouraging and allowing their clinical application.
The best cryopreservation technique for bone marrow should be effective when applied to the whole tissue, with the idea that stem cells would be isolated following thawing. The purpose is to preserve clinical samples for stem-cell recovery at a later stage, because immediate cryopreservation of tissues would be more practical than direct primary isolation of stem cells, which requires further processing. 10 Indeed, as previously reported by Woods et al. on other MSC sources, isolating human BM-MSCs (hBM-MSCs) can be toilsome, time-consuming, and expensive, especially while employing current good manufacturing practice (GMP) standards for clinical use of the cells. Hence, cryopreservation of whole bone tissue samples (including the bone marrow) or isolated bone marrow may be favorable for the banking of specimens from which BM-MSC cultures are not immediately needed. If cells are immediately needed from tissues, cryopreservation procedures need to be as efficient as possible to optimize the usefulness of the stem cells. 11 Although there are several investigations on cryopreservation of hBM-MSCs, 12 to the best of the authors' knowledge, a study that combines the use of both human serum (HuS) and low concentrations of cryoprotectants is lacking. A previous study demonstrated that HuS represents an appropriate supplement for the in vitro expansion of human dental pulp stem cells (DPSCs) by promoting the proliferation rate in comparison with cells maintained in culture with fetal calf serum or in serum-free medium. Furthermore, HuS did not induce cell senescence or apoptosis, and it did not affect the stemness of DPSCs. 13 Based on these results, the aim of the present study was to develop a novel cryopreservation technique using autologous HuS that would allow for long-term storage of human bone-marrow tissue without toxicity, and without affecting the characteristic of the hBM-MSCs derived from the frozen tissue. The use of HuS for the cryopreservation and subsequent expansion of hBM-MSCs meets the criteria approved by the committee for medicinal products for human use, and could be applied in human cell therapy with GMP protocols. 14
Materials and Methods
HuS collection
HuS was obtained from the blood of healthy male volunteers collected after provision of informed consent. The whole blood was processed as previously described by Pisciotta et al. 13 Briefly, after collection into 8-mL tubes containing silica beads for clot activation (Greiner Bio-one, Kremsmünster, Austria), the whole blood was stored at room temperature for 3 h and centrifuged at 1,400g for 15 min in order to separate erythrocytes and coagulum contents. The collected serum was heat inactivated at 56°C for 30 min, then filtered with a 0.22-μm syringe-driven filter unit (Millipore, Billerica, MA). Aliquots of sterile HuS were stored at −20°C.
Human bone-marrow stem-cell isolation
Fresh human bone specimens were obtained from the femoral heads of male patients between 18 and 21 years of age undergoing total orthopedic surgical procedures. Informed consent was obtained according to Italian law and the guidelines of the ethical committee of IRCCS Arcispedale Santa Maria Nuova, Reggio Emilia, Italy. Bone tissue consisting mainly of trabecular elements dissociated from the cortical bone was reduced to small fragments (<1 mm3) mechanically using bone nippers. Part of the bone fragments (non-frozen control cells) were incubated in culture medium (α-Minimum Essential Medium [α-MEM] +10% HuS +2 mM L-glutamine, 100 IU/mL penicillin, 100 μg/mL streptomycin), while the rest of the fragments were stored in cryopreservation solution (α-MEM +40% HuS +5% dimethyl sulfoxide [DMSO]); all bone-marrow fragments were stored in liquid nitrogen. Bone fragments were frozen overnight in a Nalgene Cryo 1°C “Mr. Frosty” Freezing Container (Thermo Scientific), using a cooling rate of 1°C/min from room temperature (RT) to −80°C, transferred to liquid nitrogen, and stored at −196°C for up to 1 year. Fresh bone fragments and the cryopreserved fragments were maintained for 2 days in flasks containing culture medium. Bone fragments were removed, and adherent cells derived from the fragments were kept in standard culture conditions (5% CO2 in air, 37°C, high humidity) with the medium changed every 3–4 days.
Immunophenotypic characterization
Cells derived from either fresh or cryopreserved bone fragments were sub-cultured until reaching 80% confluence. Following trypsin dissociation, cells were re-suspended in culture medium and confirmed, using flow cytometry, to meet the minimal criteria to be defined as MSCs, according to the standards established by the International Society for Cellular Therapy. 15 Cells were stained with the following monoclonal antibodies (mAbs): anti-human-CD73-PE-CY7, -CD90-FITC, -CD105-APC, -CD16-FITC, -CD45-PE, and -HLA-DR-PECY7 (all BD Biosciences); and -CD34-ECD (Beckman Coulter). Cells were incubated for 20 min at room temperature and washed with Stain Buffer (BD Bioscience). A minimum of 10,000 cells per sample was acquired and analyzed using the Attune Acoustic Focusing Flow Cytometer. Data were analyzed by FlowJo 9.5.7 (Treestar, Inc., Ashland, OR) under MacOS 10. The expression of MSC markers was confirmed after 6 passages.
Cell proliferation and colony-forming fibroblast assays
The proliferation rates of non-frozen, control cells obtained from fresh fragments and of cells obtained from fragments cryopreserved and stored for 1 year at −196°C were analyzed. After reaching confluence, adherent cells, isolated as described above, were dissociated and seeded in 60-mm Petri dishes at a density of 4 × 103 cells/cm2, and cultured for 1 week until reaching confluence. Cell counting was performed each day on 6 randomly selected fields of 1 mm2 (20× magnification) for each dish (3 different samples for each experimental group) using a Nikon TE2000 inverted microscope. All analyses were performed by individuals blinded to the experimental details. Cell density was expressed as the mean of cells/cm2 ± standard deviation (SD). The population doubling (PD) time was calculated in the exponential growth phase using the following formula:
where N7d is the cell number at day 7 and N1d is the cell number at day 1. To determine the PD rate, hBM-MSCs were initially seeded at a density of 4 × 103 cells/cm2 in culture media. Cells were harvested and counted once they reached 80% confluence. At each passage, cells were re-plated at the initial density, and cultures were performed until passage 5. The following formula was applied to 3 samples for each experimental group:
where N is the harvested cell number and Ns is the initial number of cells plated. A cumulative population doubling (CPD) index for each passage was obtained by adding the PD of each passage to the PD of the previous passages. In order to evaluate the clonogenic potential, a colony-forming fibroblast (CFU-F) assay was performed. Cells from passages 1 and 6, isolated from fresh fragments and from fragments cryopreserved for 1 year, were plated at 65 cells/cm2 in 12-well polystyrene tissue culture plates and stored in basal culture medium for 14 days at 37°C in a 5% CO2 humidified atmosphere. The medium was then removed, and the cells were fixed with methanol/acetic acid 3:1 for 5 min, and stained with 0.5% crystal violet in methanol for 30 min at room temperature. A positive CFU-F colony was identified as an adherent colony containing at least 50 cells and was visualized at 10× magnification using a Nikon TE2000 inverted microscope. At the same time, a limiting dilution assay was performed, plating the cells at 125, 65, 32, and 16 cells/cm2 in a 12-well plate under the CFU-F assay conditions described above. After 2 weeks, the number of CFU-F colonies generated was counted.
Karyotype analysis and p53 expression
Cells isolated from fresh and cryopreserved fragments were treated with 30 ng/mL of Colcemid Solution (Invitrogen) for 3 h to arrest the cells in metaphase. The cells were then harvested, resuspended in a hypotonic solution (KCl 75 mM), and incubated at 37°C for 12 min. Cells were then centrifuged and fixed in cold methanol:acetic acid (3:1; v/v). After fixation, the cells were spread onto glass slides, allowed to dry, and stained with Giemsa Stain (Invitrogen). After washing the slides with water, a minimum of 10 chromosome metaphases were counted, and the modal number recorded.
The expression of p53 was evaluated by immunofluorescence and Western blot analysis. hBM-MSCs cryopreserved with standard solution consisting of 40% fetal bovine serum (FBS) and 10% DMSO were used as control.
Senescence and cell death
In order to evaluate the presence of senescent cells in hBM-MSCs obtained from both fresh and cryopreserved fragments, cells at passage 6 were seeded in 12-well plates and processed using a senescence β-Galactosidase staining kit (Cell Signaling), according to the manufacturer's instructions. Three samples for each culture condition were analyzed, and the percentage of senescent cells calculated. The apoptosis process was evaluated by Western blot analysis, assessing the expression of the cleaved form of caspase 3. Whole-cell lysates of hBM-MSCs obtained from both fresh fragments and cryopreserved fragments at passages 1 and 6 were processed as previously described, 16 and caspase 3 was detected using an anti-caspase-3-specific Ab (Santa Cruz Biotechnology, Inc.). A sample of lymphocytes treated with 1 μM of Staurosporine (Sigma-Aldrich) was used as positive control, while a sample of fibroblasts was loaded as negative control.
Cell differentiation assays
Cells obtained from both fresh fragments and fragments cryopreserved for 1 year at passage 1 were tested for their ability to differentiate toward osteogenic, adipogenic, and myogenic lineages. For each differentiation experiment, 3 samples per condition were used.
Osteogenic differentiation
Osteogenic differentiation was carried out as previously described by Jaiswal et al. 17 Briefly, cells were seeded at approximately 3 × 103 cells/cm2 on culture dishes and kept in culture medium until reaching confluence The medium was then replaced by osteogenic medium (culture medium supplemented with 100 μM 2P-ascorbic acid, 100 nM dexamethasone, 10 mM β-glycerophosphate) containing 5% HuS. The medium was changed twice a week. After 3 weeks of osteogenic induction, alkaline phosphatase activity, extracellular calcium deposition, and the expression of osteogenic markers were evaluated.
Adipogenic differentiation
Cells were incubated for 3 weeks in adipogenic induction medium according to Pittenger et al. 18 (culture medium supplemented with 0.5 mM isobutyl-methylxanthine, 1 μM dexamethasone, 10 μM insulin, 200 μM indomethacin, and 10% HuS). Medium was changed every 3 days. Lipid-rich vacuoles within the cells were evaluated by Oil red O staining and AdipoRed assay (LONZA), following the manufacturer's instructions.
Myogenic differentiation
Experiments were carried out according to Pisciotta et al. 13 In order to investigate the capability of hBM-MSCs to participate in myotube formation, cells were seeded at direct co-culture with C2C12 mouse myoblast cells. hBM-MSCs and C2C12 cells were seeded in a 10:1 ratio in DMEM High Glucose, supplemented with 10% HuS, 2 mM L-glutamine, 100 IU/mL penicillin, and 100 mg/mL streptomycin, until confluence was reached. Growth medium was then replaced with DMEM High Glucose supplemented with 1% HuS and 10 nM insulin. Cells were maintained in co-culture for 2 weeks. Double immunofluorescence staining using anti-human Nuclei (hNu) ab and anti-desmin was performed in order to verify the formation of myotubes with the direct contribution of hBM-MSCs.
Histochemistry and Western blotting
Some samples of in vitro differentiated hBM-MSCs were fixed in 4% paraformaldehyde in phosphate buffered saline (PBS) at pH 7.4 for 20 min and processed for subsequent analysis. For Alizarin red staining, fixed cells were incubated for 5 min at room temperature in a solution containing 0.1% Alizarin red and 1% ammonium hydroxide. Other samples were fixed in methanol/acetone 3:7 for 10 min at 22°C in order to perform the alkaline phosphatase assay. Fixed cells were processed using the Leukocyte Alkaline Phosphatase Kit (Sigma-Aldrich), following the manufacturer's instructions. Images were collected using a CCD color camera equipped with a 90-mm macro photograph objective.
Densitometry was performed on culture plates from 3 independent experiments using NIS software (Nikon). An equal area (region of interest) was selected around the plate surface, and the mean of gray levels (in a 0–256 scale) was calculated. Data were then normalized to values of background and expressed as mean ± standard deviation (SD).
For Western blot analysis, whole-cell lysates were obtained from undifferentiated and differentiated hBM-MSCs isolated from fresh fragments and fragments cryopreserved for 1 year and processed as described by Gibellini et al. 19 Forty micrograms of total protein from each sample was separated by SDS-PAGE and then transferred to polyvinylidene difluoride membranes. Blots were incubated with primary Abs mouse anti-OCN (Abcam); mouse anti-human type I collagen (Coll-I; Millipore); rabbit anti-caspase 3, Active (Sigma-Aldrich), diluted 1:1,000, and revealed by horseradish peroxidase-conjugated secondary Abs (anti-mouse; anti-rabbit; Pierce Antibodies, Thermo Scientific) diluted 1:3,000. All membranes were revealed using Enhanced Chemiluminescence (Amersham). Anti-actin Ab was used as control of protein loading in timing experiments. Densitometry was performed on 3 independent experiments using NIS software. An equal area was selected inside each band, and the mean of grey level (0–256 scale) was calculated. Data were then normalized to values of background and an actin band control.
Immunofluorescence and confocal microscopy
Monolayer cells were fixed in 4% ice-cold paraformaldehyde in PBS for 20 min, permeabilized with 0.1% Triton X-100 in PBS for 5 min, and then processed as previously described. 20 The following primary Abs diluted 1:100 were used: mouse anti-human nuclei (hNu; Millipore]; mouse anti-OCN (Abcam); rabbit anti-osterix (OSX; GeneTex); rabbit anti-myosin, rabbit anti-desmin (Sigma-Aldrich); mouse anti-human p53 (Santa Cruz Biotechnology, Inc.). Secondary Abs (goat anti-mouse Alexa488, goat anti-rabbit Alexa546, goat anti-mouse Alexa546; Invitrogen) were diluted 1:200. Nuclei were stained with 1 μg/mL 4′,6-diamidino-2-phenylindole (DAPI) in PBS. Negative controls consisted of samples not incubated with the primary Ab. The multi-labeling immunofluorescence experiments were carried out avoiding cross-reactions between primary and secondary Abs.
Confocal imaging was performed using a Nikon A1 confocal laser scanning microscope, as previously described. 21 The confocal serial sections were processed with ImageJ software to obtain 3-dimensional projections and image rendering was performed by Adobe Photoshop Software.
Data analysis
Values are reported as the mean ± SD obtained with groups of 5 samples each. Differences between 2 experimental samples were analyzed by paired Student's t-test. Three or more experimental samples were analyzed by analysis of variance (ANOVA) t-test followed by Dunnett's test (GraphPad Prism Software, Inc., v5). In all cases, significance was set at P < 0.05.
Results
Isolation and growth kinetics of hBM-MSCs
Fresh and cryopreserved fragments (<1 mm3) were incubated in basal culture medium supplemented with HuS (Fig. 1). During culture, adherent cells were observed in the culture dish after 2 days of incubation. Trabecular fragments were removed after 2 days; cells reached 80%–90% of confluence after 2 weeks of culture, changing the medium twice a week. Bone-marrow cells gradually generated a sub-confluent layer of fibroblast-like, spindle-shaped cells. This kind of morphology displayed by both hBM-MSC populations was observed by inverted light microscope through the whole culture time, as shown in Fig. 2A, and no differences were observed between the 2 experimental groups. The first passage was performed after 14 days, and the expression of MSC surface antigens was evaluated by fluorescence-activated cell sorting (FACS) analysis. Cells were re-seeded, and the growth kinetics analyzed. The growth rate of hBM-MSCs isolated from fresh and cryopreserved fragments was evaluated during routine cell culture conditions, and cell counting was performed up to day 7, until confluence was reached.
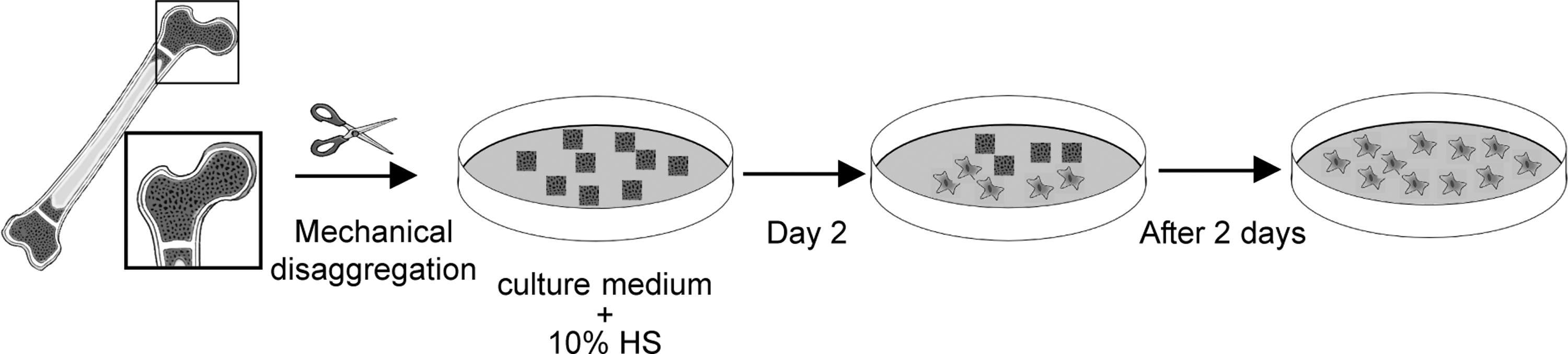
Isolation of human bone-marrow mesenchymal stem cells (hBM-MSCs). Schematic representation of the processing of trabecular bone enclosing bone marrow. Bone fragments obtained after mechanical disaggregation were divided into 2 parts, with 1 part maintained in culture with human serum (HuS), while another part underwent cryopreservation.
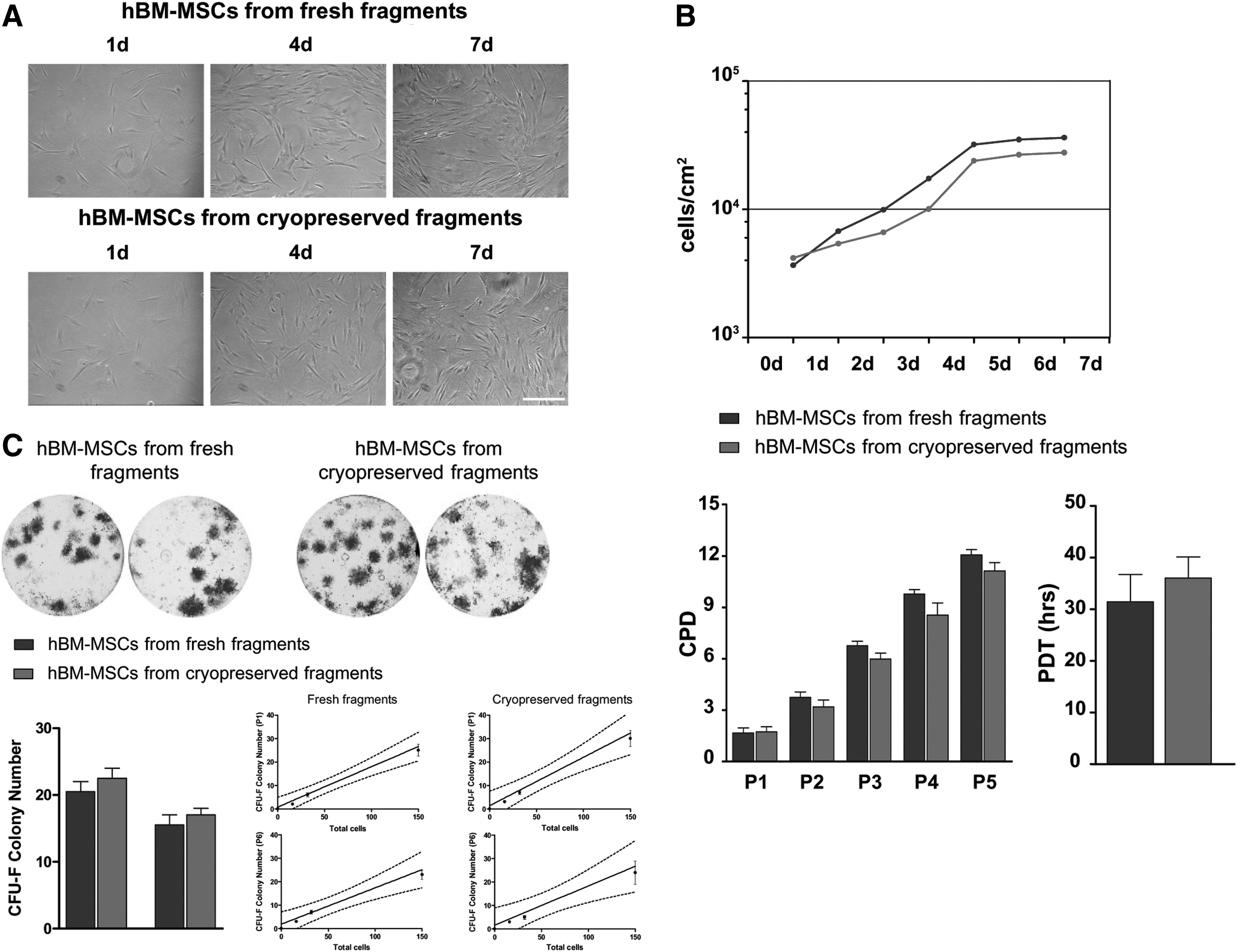
Cell morphology and proliferation. (
hBM-MSCs isolated from cryopreserved fragments and cultured with HuS showed a slightly lower proliferation rate compared with the hBM-MSCs isolated from fresh fragments, although the difference was not significant (Fig. 2B). This slight difference was found by analyzing the PDT of both isolated hBM-MSC populations (from fresh fragments: 31.4 ± 5.3 h; from cryopreserved fragments: 36.0 ± 4.1 h). Similar results were obtained in the CPD analysis. As shown in Fig. 2B, at each passage, hBM-MSCs from fresh fragments proliferated slightly faster compared to the hBM-MSCs from cryopreserved fragments. In all cases, the ANOVA t-test did not show statistically significant differences.
CFU ability
hBM-MSCs isolated from fresh fragments and from cryopreserved fragments at passages 1 and 6 were analyzed by assaying for colony-forming potential. As shown in Fig. 2C, cells obtained from both conditions were capable of forming CFU-F at passages 1 and 6. Statistical analysis did not reveal any significant differences between the 2 experimental groups.
With regard to the limiting dilution assay, the number of CFU-F formed was linearly related to the number of hBM-MSCs plated. This ability was observed in both experimental groups at passages 1 and 6. The linear relationship between the number of CFU-F and number of hBM-MSCs seeded from fresh fragments at passages 1 and 6 was R2 = 0.95 and R2 = 0.90, respectively. A linear relationship between the number of CFU-F and cell number was also observed in cells isolated from cryopreserved fragments at passages 1 and 6 (R2 = 0.92 and R2 = 0.84, respectively; Fig. 2C, bottom, left side).
Immunophenotypic characterization
In order to evaluate the expression of MSC surface antigens, hBM-MSCs isolated from fresh fragments and from those stored for 1 year were analyzed by flow cytometric analysis (Fig. 3). At passage 1, both groups of hBM-MSCs were positive for CD73 (98.3% and 99.5%), CD90 (99.8% and 98.6%), and CD105 (99.9% and 99.1%) mesenchymal cell surface antigens. Conversely, cells isolated from both fresh and cryopreserved fragments did not express the hematopoietic and endothelial markers CD16 (99.9% and 99.9%), CD34 (99.9% and 99.6%), CD45 (99.9% and 99.9%), and the class II Major Histocompatibility Complex (MHC) cell surface receptor HLA-DR (99.9% and 99.3%). No differences were observed between the 2 experimental groups (freshly isolated and cryopreserved MSCs).
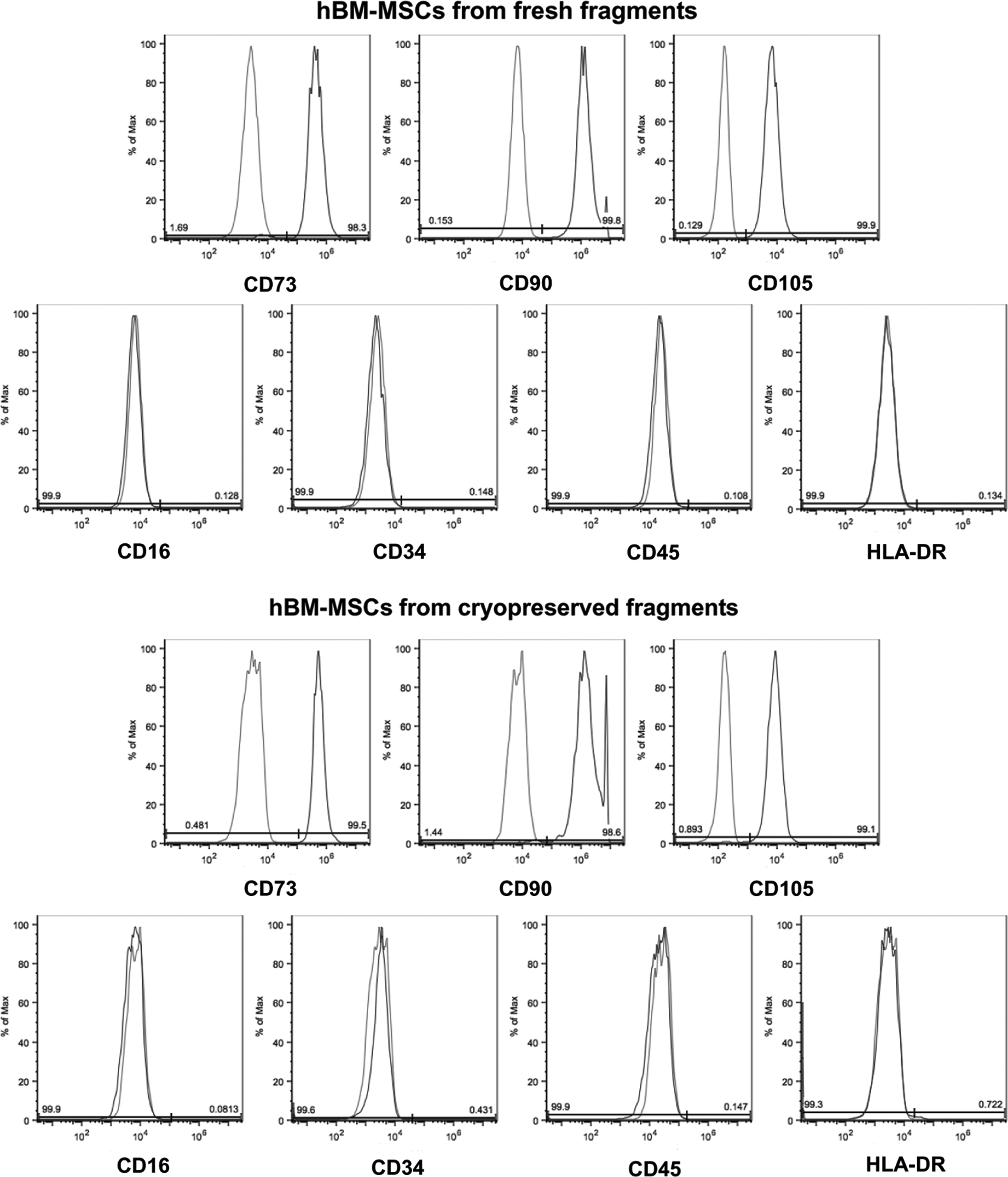
Immunophenotypic profile. Fluorescence-activated cell sorting (FACS) analysis was carried out on hBM-MSCs isolated from fresh fragments (top) and from those stored for 1 year (bottom).
In order to investigate whether cells derived from cryopreserved fragments maintained mesenchymal characteristics until late passage, they were cultured upon passage 6, tested for surface marker expression, and compared with hBM-MSCs isolated from both fresh and cryopreserved fragments at passage 1. FACS analysis confirmed that hBM-MSCs obtained from cryopreserved fragments at passage 6 maintained the expression of specific MSC markers (CD73, CD90 and CD105); no expression of CD16, CD34, CD45, and HLA-DR was seen, similar to that for hBM-MSCs isolated from fresh and cryopreserved fragments at passage 1 (Fig. 4A). Similar results were observed by FACS analysis carried out on hBM-MSCs from fresh fragments at passage 6 (data not shown).
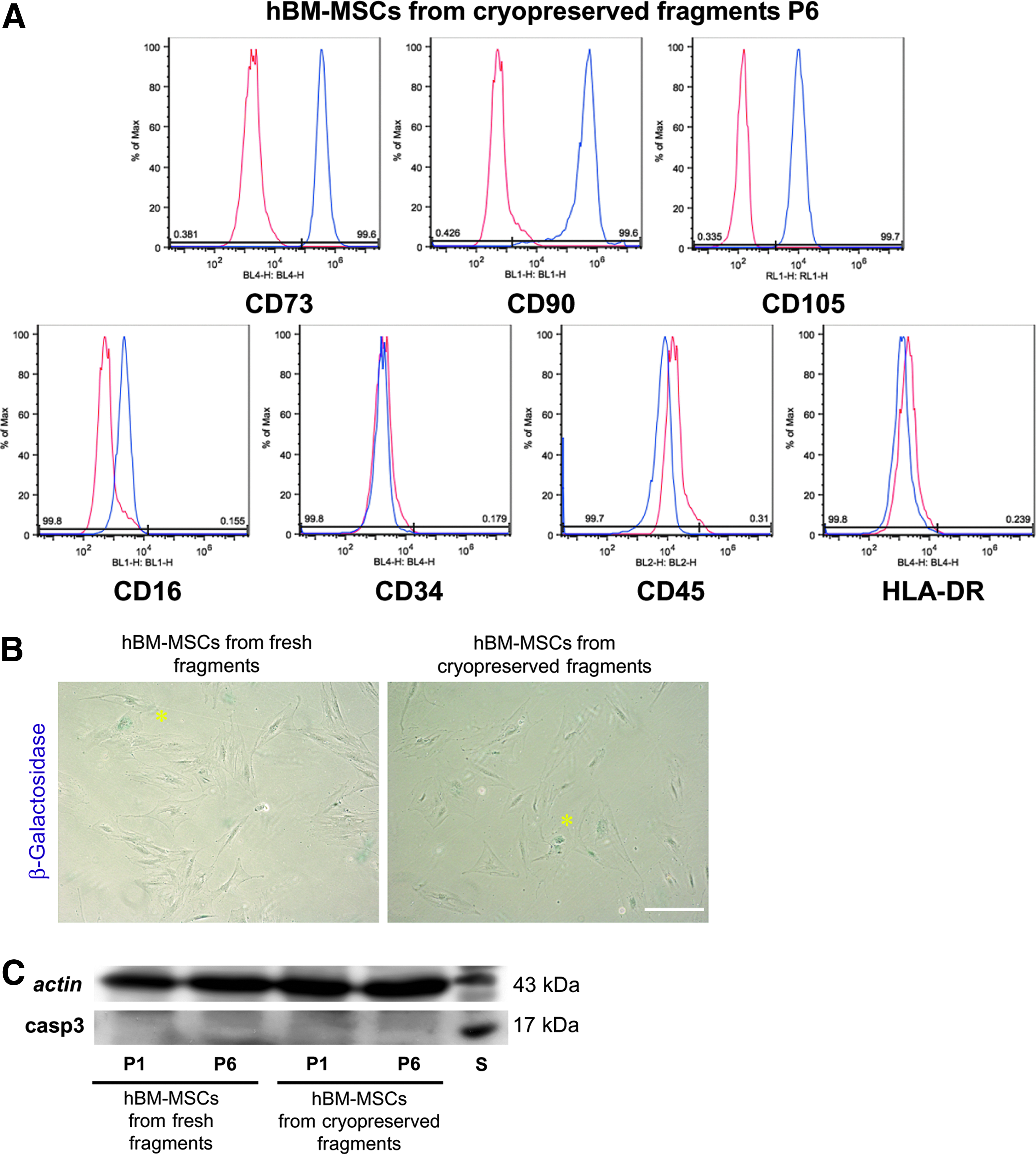
Mesenchymal cell surface antigens, senescence, and cell death. (
Senescence and apoptosis
Cell senescence was evaluated by detection of β-galactosidase in cultures of hBM-MSCs isolated from fresh and cryopreserved fragments at passage 6. Very low levels of β-galactosidase activity were detected in both hBM-MSC cultures (Fig. 4B, arrowheads); no significant differences were observed between the 2 hBM-MSC cultures in the percentage of senescent cells detected by microscopic observation (hBM-MSCs from fresh fragments 0.064% ± 0.007%; hBM-MSCs from cryopreserved fragments 0.059% ± 0.004%; n = 3). In order to evaluate the apoptosis occurring in hBM-MSCs from both experimental groups at passages 1 and 6, Western blot analysis of active caspase 3 was carried out in whole-cell lysates obtained from both hBM-MSC cultures at passages 1 and 6. Very low expression of active caspase 3 (casp3; 17 kDa) was observed at both passages 1 and 6 in the 2 experimental groups. On the other hand, the positive control consisting of lymphocytes treated with 1 μM of Staurosporine (S) showed a clear band corresponding to caspase 3, which was not appreciable in either hBM-MSC cultures (Fig. 4C). Densitometric analysis performed on the caspase 3 bands did not show any significant differences between hBM-MSCs isolated from fresh and cryopreserved fragments at passages 1 and 6. These data indicate that the cryopreservation of trabecular fragments for 1 year and the subsequent isolation of hBM-MSCs, as previously described, did not induce premature senescence and did not affect the survival of hBM-MSCs themselves.
Karyotype analysis and p53 expression
hBM-MSCs isolated from fragments cryopreserved (CF) for 1 year showed a normal karyotype similar to the cells isolated from fresh fragments (FF) and those cryopreserved with the standard solution (STD; Fig. 5A). Moreover, no significant differences were observed in the expression of p53 at passages 1 and 6 (Fig. 5B). These data suggest that the cells entrapped in the bone fragments under prolonged cryopreservation maintained their genomic stability without showing DNA damage.

Karyotype analysis and p53 expression. (
Multi-lineage differentiation capacity
An effective cryopreservation protocol for the storage of trabecular fragments enclosing the bone marrow should not only allow for isolation of bone-marrow-derived stem cells at a later time point, but should also not cause any abnormalities in biological function. For this purpose, the ability of hBM-MSCs isolated from fresh fragments and fragments cryopreserved for 1 year to differentiate in vitro along osteogenic, adipogenic, and myogenic lineages were assessed and compared.
Osteogenic differentiation
Alkaline phosphatase (ALP) is considered a routine marker for osteogenic differentiation of MSCs.17,18 Normally, intracellular ALP activity readily increases during in vitro osteogenesis, forming violet, insoluble granular dye deposits. After 4 weeks of differentiation, cell cultures from both fresh and cryopreserved fragments were intensely positive for ALP, extending to the whole culture plate. No significant differences were observed between the 2 experimental groups following densitometric analysis. Moreover, hBM-MSC cultures from both fresh and cryopreserved fragments started to form nodular aggregates after 10 days of induction in osteogenic medium. After 4 weeks, mineral deposits appeared in the extracellular space and in nodular aggregates (data not shown). The in vitro deposition of mineralized extracellular matrix was analyzed by Alizarin red staining. As confirmed by densitometric analysis, a large amount of mineralized extracellular matrix was detected in differentiated hBM-MSCs isolated from both cultures, and no differences were observed between them. Undifferentiated cells were negative for both Alizarin red and ALP (Fig. 6A). Confocal analysis at day 28 showed the expression of OCN and OSX, another specific marker of osteogenic commitment. Double immunofluorescence staining was carried out to analyze the localization of OCN and OSX at the same time in hBM-MSCs from fresh and cryopreserved fragments (Fig. 6B). OCN was typically localized in the cytoplasm with strong expression in both experimental groups, while OSX showed a peculiar nucleoplasmic localization (Fig. 6B). In order to confirm the osteogenic induction of hBM-MSCs isolated from fresh and cryopreserved fragments, the expression of Collagen I and OCN was evaluated by Western blot analysis (Fig. 6C): Collagen I and OCN were detected in hBM-MSCs from both experimental groups, while undifferentiated samples displayed no expression for either marker.
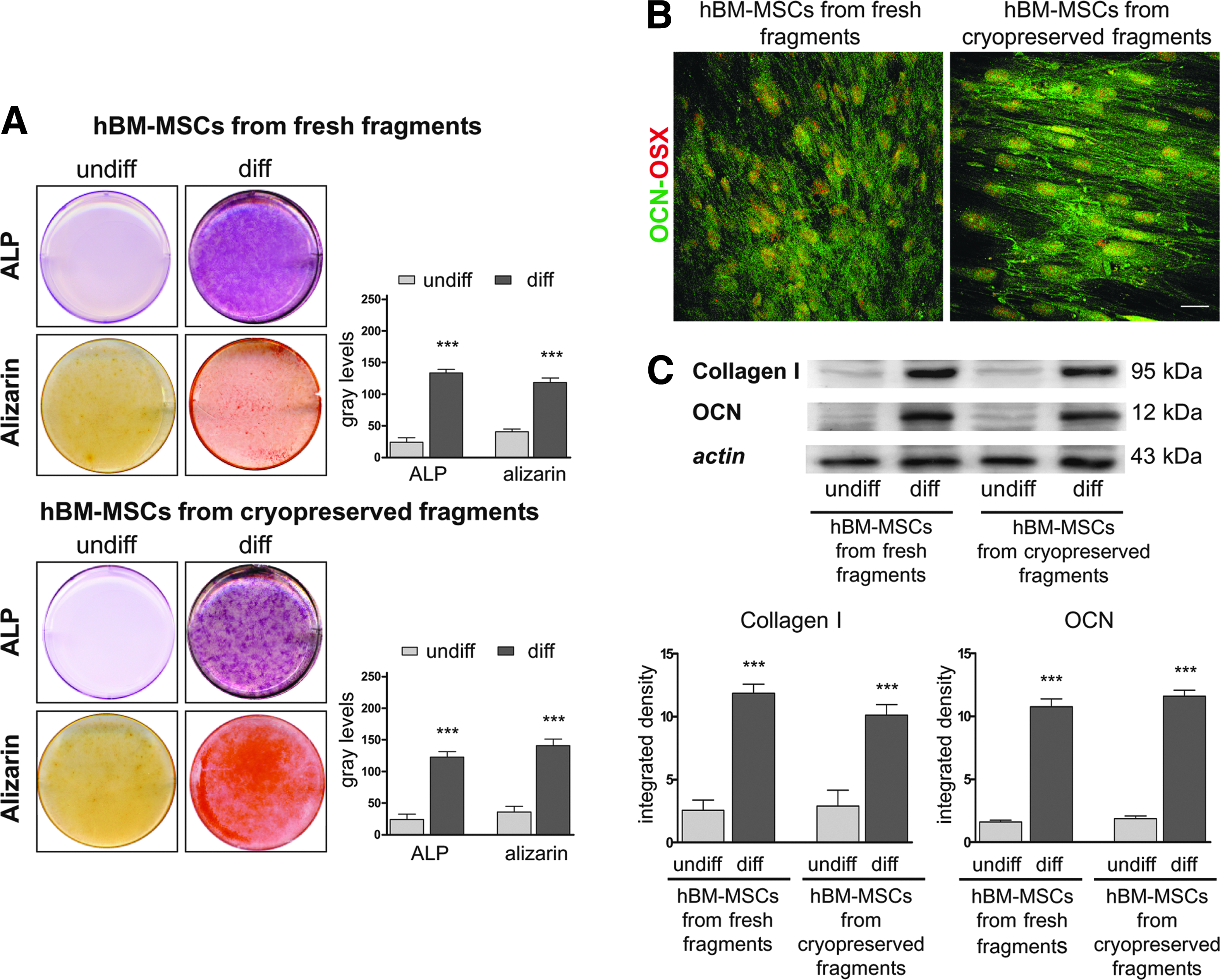
Evaluation of osteogenic differentiation. (
Adipogenic differentiation
hBM-MSCs isolated from fresh and cryopreserved fragments were cultured in adipogenic medium for a total of 3 weeks. Both hBM-MSC cultures started to form intracellular lipid drops as early as day 5 of differentiation. After 3 weeks, the morphological changes in hBM-MSCs isolated from fresh and cryopreserved fragments were evident, and characterized by the accumulation of numerous lipid drops in the cytoplasm clearly stained by Oil red O (Fig. 7A). The lipid drops and the quantitative measurement of intracellular lipids were confirmed using the AdipoRed™ assay. As shown at the top of Fig. 7B, the immunofluorescence images obtained after the incubation of differentiated hBM-MSCs isolated from fresh and cryopreserved fragments using AdipoRed™ showed the accumulation of lipid droplets in the cytoplasm. The lipid contents of the differentiated and undifferentiated groups were compared (Fig. 7B). A comparison of relative fluorescence indicated that while there is no significant difference in lipid content between hBM-MSCs isolated from fresh and cryopreserved fragments after 3 weeks of differentiation, there was a significant difference between lipid content between differentiated and undifferentiated hBM-MSCs (Fig. 7B).
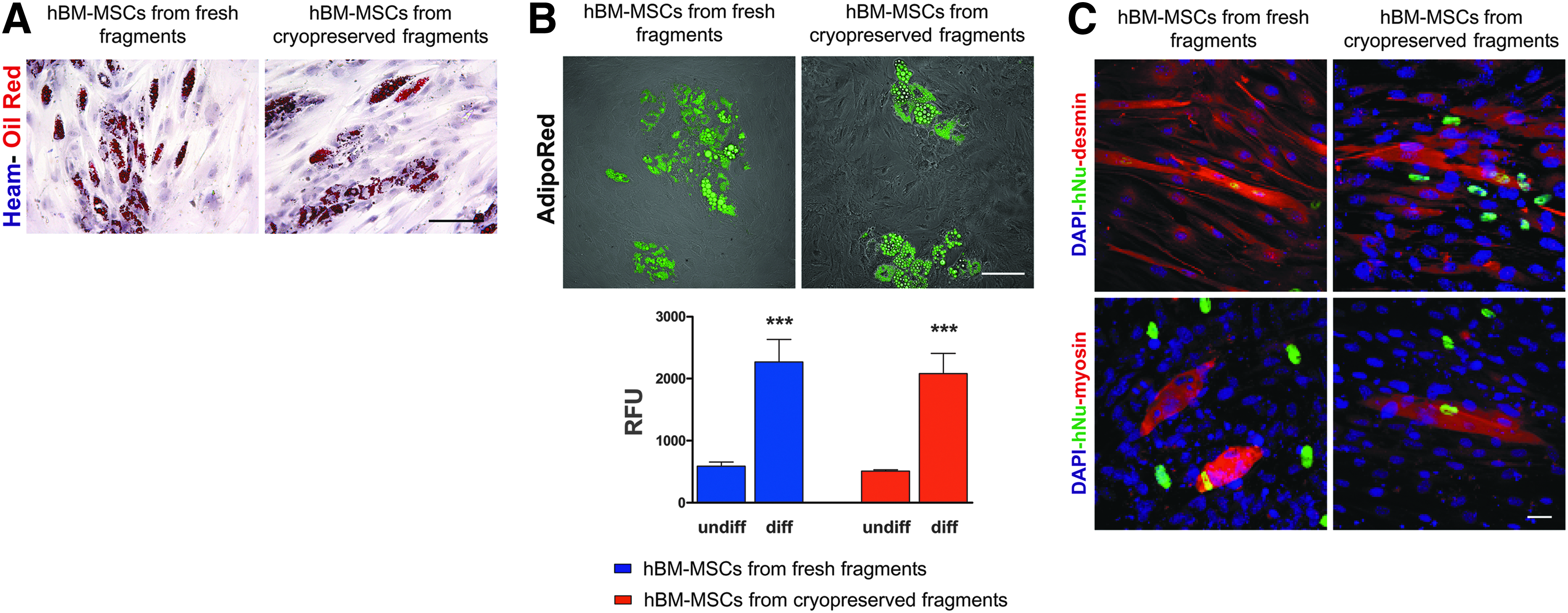
Evaluation of adipogenic and myogenic differentiation. (
Myogenic differentiation
The ability of hBM-MSCs isolated from fresh and cryopreserved fragments to differentiate toward myogenic lineage was verified by direct co-culture with C2C12 mouse myoblasts. After 2 weeks, myotube formation was observed in both co-cultures. Myotubes appeared multi-nucleated, indicating that cell fusion occurred. Labeling by anti-human nuclei antibody (anti-hNu) demonstrated that hBM-BMCs were involved in myotube generation (Fig. 7C). Double staining with anti-hNu/anti-desmin antibodies and anti-hNu/anti-myosin antibodies indicated that hBM-MSCs isolated from both fresh and cryopreserved fragments were able to form mature hybrid myotubes. Myotubes not labeled by anti-hNu antibody and therefore formed only by C2C12 cells were also present (Fig. 7C). The percentage of hybrid myotubes (formed with the contribution of hBM-MSCs) respect to the total myotubes, was similar in both experimental groups (co-culture of C2C12 with hBM-MSCs from fresh fragments 42.7% ± 6.4%; co-culture of C2C12 with hBM-MSCs from cryopreserved fragments 46.3% ± 7.2%; n = 4).
Discussion
MSCs, also referred to as multipotent mesenchymal stromal cells, were originally isolated from bone marrow, and have subsequently been retrieved from many other organs and tissues.2,3,18,22,23 Despite enormous progress and an increased understanding of MSCs at the molecular and cellular levels, several critical questions remain with regard to the use of these cells in therapeutic applications. On a clinical level, both autologous and allogenic approaches for the transplantation of MSCs are being investigated. Most of the processing phases required for the clinical application of MSCs include isolation from various tissues, in vitro expansion, and especially cell banking. A major challenge in the banking of MSCs is the development of a cryopreservation protocol that is compliant with the European regulations on advanced therapy and with GMP standards, and allows long-term storage without affecting the characteristics of the cells. The most widely used method for banking of hBM-MSCs consists of: (1) isolation according to the criteria established by Friedenstein et al., 24 and further modified by others,25,26 based on the physical propensity of MSCs to adhere to plastic flasks; and (2) in vitro expansion and subsequent storage in liquid nitrogen.
The performance of this protocol in accordance with GMP would require a long time and would be difficult and extremely expensive, and the contamination risk is very high. An additional factor that must be considered for cell banking is the need for improvement of cryopreservation protocols. Currently, cells are typically preserved in a freezing medium consisting of Dulbecco's modified Eagle's medium (DMEM) or α-MEM, FBS, and DMSO. Caution with the use of FBS in clinical settings has been suggested because of potential disease transmission and xenogenic immunologic response. 27 Previous work indicates that FBS proteins can be internalized in stem cells and cause pathogen transmission; bovine proteins may also be identified as antigenic substrates leading to a xenogenic immune response.28,29 For these reasons, in addition to use of GMP, clinical protocols for cell therapy encourage the use of FBS substitutes. 30 Serum-free cryopreservation media have been tested; they failed and did not constitute adequate support for proliferation and viability of MSCs.31–33 In this regard, it would be reasonable to assume that cryopreservation of whole bone tissue samples (including the bone marrow) using HuS for subsequent stem-cell recovery may be advantageous for the banking of specimens from which BM-MSC cultures are not immediately needed. If cells are immediately needed from tissues, cryopreservation procedures need to be as functional as possible to maximize the usefulness of the stem cells. In addition, the use of HuS for cryopreservation that belongs to the same donor who will benefit from the stem cells would represent an enormous therapeutic advantage, thus encouraging the autologous clinical application of hBM-MSCs.
This study describes a straightforward protocol for the cryopreservation of trabecular fragments containing bone marrow. After cryopreservation in a medium supplemented with 5% DMSO and 40% HuS, the biological properties of hBM-MSCs isolated from the fragments were thoroughly examined. The efficiency of the protocol was assessed by growth kinetics, surface marker expression, and the in vitro differentiation potential of the cells, and their biological activity was compared with the hBM-MSCs isolated from fresh fragments.
The data presented here demonstrate that the isolation of adherent hBM-MSCs from cryopreserved fragments gives rise to a morphologically homogeneous population of cells that maintain a good proliferation rate during culture, and exhibit growth kinetics that are comparable to the counterpart isolated from fresh fragments.
Based on morphological features and proliferative activity, most of the adherent cells in culture are MSCs (fibroblast-like), excluding the possible migration of osteocytes (polygon in shape), according to Zhu et al. 34 Moreover, prior to and following cryopreservation, hBM-MSCs isolated from trabecular bone fragments were capable of giving rise to fibroblastoid colonies (CFU-F), and the linear relationship between colony number and seeded cells was maintained at both early and late passages. Based on these properties, the cells isolated from fresh and cryopreserved fragments reflect the peculiarities of MSCs, as established by Friedenstein et al. 24 To identify the adherent cells, immunophenotypic and differential features were analyzed. In summary, the cell surface antigen profile showed that the cultured hMSCs belong to a population of non-hematopoietic, non-endothelial, but mesenchyme-derived adult stem cells. Their immunophenotypic profile was maintained throughout the entire culture period, up to passage 6, and no significant induction to premature cell senescence and/or death was observed. Moreover, no karyotype alterations were observed or any DNA damage detected.
However, characterization based on surface marker expression alone is not sufficient to define MSCs.35,36 Therefore, multi-lineage differentiation assays were performed to identify whether the cells grown out from bone fragments were MSCs. The data demonstrate that cells isolated from both fresh and cryopreserved fragments and cultured in induction media were able to differentiate toward osteogenic, adipogenic, and myogenic lineages. In conclusion, these results imply that minimal tissue processing may be adequate for the banking of samples with no immediate plans for hBM-MSC expansion and use, which in turn may limit cost and contamination risk, and facilitate the banking for future clinical use. The use of HuS in the protocol for cryopreservation of trabecular fragments as well as for expansion and differentiation of hBM-MSCs allows for clinical application of cells with GMP-compatible protocols, consistent with the criteria approved by the World Health Organization.
Footnotes
Acknowledgments
This work was supported by grants from “Progetto Strategico per lo sviluppo nella sede di Reggio Emilia della Facoltà di Medicina e Chirurgia” Prot: 2010 0007725, Arcispedale S. Maria Nuova di Reggio Emilia and MIUR FIRB Accordi di Programma 2010 Prot: RBAP10Z7FS.
Author Disclosure Statement
No competing financial interests exist.