
Abstract
A high survival rate of cryopreserved cells requires optimal cooling and thawing rates in the presence of a cryoprotective agent (CPA) or a combination of CPAs in adequate concentrations. One of the most widely used CPAs, dimethyl sulfoxide (Me2SO), however is toxic at high concentrations and has detrimental effects on cellular functions. Additional processing steps are necessary to remove the CPA after thawing, which make the process expensive and time consuming. Therefore it is of great interest to develop new cryoprotective strategies to replace the currently used CPAs or to reduce their concentration. The aim of this study was to investigate if thermal activation of human pulmonary microvascular endothelial cells (HPMEC ST-1.6R), prior to cryopreservation, could improve their post-thaw viability since the resulting heat shock protein expression acts as an intrinsic cellular protection mechanism. The results of this study suggest that both heat and cold shock pretreatments improve cryopreservation outcome of the HPMEC ST-1.6R cells. By re-cultivating cells after heat shock treatment before cryopreservation, a significant increase in cellular membrane integrity and adherence capacity could be achieved. However a combination of thermal activation and cryopreservation with alternative CPAs such as ectoine and L-proline could not further enhance the cell viability. The results of this study showed that pretreatment of endothelial cells with thermal activation could be used to reduce the Me2SO concentration required in order to preserve cell viability after cryopreservation.
Introduction
T
The induction of intrinsic cellular protection mechanisms such as the heat shock proteins offer a promising alternative. Heat shock proteins (HSPs) were discovered in 1962 by Ritossa 4 and subsequently described by Tissieres in 1974. 5 To date, there are more than 11 different HSP families. The nomenclature of HSPs is based on their molecular weight, for example, HSP100, HSP90, HSP70, and small HSPs. 6 The type and number of expressed HSPs varies considerably in different organisms and cell types. In general they may be classified into two major categories: (a) 68-110 kDa and (b) 15-30 kDa. 7
These proteins are highly conserved, vital components of cellular stress response. They function as “molecular chaperones” 8 by binding to other cellular proteins, thereby ameliorating the detrimental effects of protein misfolding and promoting these proteins to return to the native structure after exposure to diverse stressful conditions. HSP expression can be induced by hyper or hypothermal stress, or on exposure to heavy metals, ethanol, and other chemicals.9–11
Previous studies have shown that overwintering insects upregulate HSP70 during diapause. 12 It was also found that the expression of HSPs enhanced the ability of pupae to survive low temperatures. 13 Russotti et al. 14 reported tolerance of human fibroblasts to hypothermal storage at 4°C after previous heat shock treatment. Similarly, Healy et al. 15 showed that renal proximal tubular epithelial cells could be protected from cold storage and re-warming injury by incubating cells at 42°C for 1 h. The group reported that cell death induced by cold storage and rewarming was associated with reduced expression of HSP70, HSP27, HSP90, and Bcl2.
Significantly reduced cell death after cold storage and re-warming by previous heat shock was associated with maintenance of HSP70, HSP27, and Bcl2 levels. Previous studies pertaining to heat shock and cryopreservation of cells have been performed.16–18 The results were controversial, but information gained from these studies emphasizes the role of heat shock proteins in cellular tolerance towards subsequent ultra low temperature stress.
Compatible solutes such as ectoine and L-proline have been previously investigated for their cryoprotective effects on various cell components and cell types.19–21 These compounds are small organic osmolytes (such as polyols, sugars, methylamines, amino acids, and some of their derivatives), which are synthesized by several microorganisms and accumulated in response to stress factors such as hyperthermia or high salinity.22,23 The incorporation of compatible solutes in freezing media offers yet another possible alternative to cryopreservation with Me2SO. A previous study from our group showed that supplementation of freezing media with ectoine and L-proline improved cell survival. 24
One aim of the current study was to investigate if thermal activation could protect human pulmonary microvascular endothelial cells from the damaging effects of cryopreservation. Endothelial cells are of great interest in transplantation and cell-based therapy and are often cryopreserved before they are cultured onto scaffolds prior to therapeutic applications.25,26 In the current study, human pulmonary microvascular endothelial cells (HPMEC ST-1.6R cell line) were pretreated with thermal activation followed by recultivation, prior to cryopreservation with various Me2SO concentrations and tested for effects on cell viability. A second set of experiments was performed to check if thermal activation in combination with compatible solutes such as ectoine and L-proline could further reduce the amount of Me2SO required to preserve cell survival after cryopreservation.
Materials and Methods
Cell culture
Human pulmonary microvascular endothelial cell line HPMEC ST-1.6R was kindly provided from J. Kirkpatrick. 27 Cells were cultured in T7 5 cm2 cell culture flasks, in Dulbecco's Modified Eagle Medium (DMEM) (Biochrom) containing 1% penicilin/streptomycin (Biochrom), 20% fetal calf serum (FCS) (Biochrom), 5105 units of heparin (Biochrom), and 10 μg/mL endothelial cell growth supplement (ECGS) (Millipore).
Hypo- and hyperthermal activation
Prior to heat or cold shock treatments, endothelial cells were cultivated until about 80% confluency. For heat shock protein induction, cells were incubated for 30 min in a CO2-incubator (Heraeus GmbH) adjusted to 43°C or a water bath adjusted to 45°C, other parameters remained unchanged. For cold shock treatment, cells were incubated in a water bath adjusted to 4°C for 30 min. After the respective pretreatments, cells were transferred to a CO2 incubator adjusted to 37°C and incubated for 4, 10, or 12 hours. Cells were then harvested for cryopreservation or pelletized by centrifugation at 235 g for 10 min at 25°C and stored at −80°C till further experimentation (RT-PCR and Western blot analysis).
Cryopreservation
Cells were detached from culture dishes with 0.25% trypsin, after which the cell number and cell viability was measured with a Vi-CELL™ cell viability analyzer (Beckman Coulter). These cells were then centrifuged, and medium was added to each cell pellet in a dropwise manner for about 10 min. Cells were then aliquoted into cryovials. Each cryovial contained 1.5 mL cell suspension with 4.5 × 105 cells/mL. The addition of freezing media to cells was performed on an ice bath. Each experiment was performed using three to six cryovials for every Me2SO concentration (0%, 1%, 2.5%, and 7,5% (v/v) Me2SO).
Freezing experiments with compatible solutes (CS) as alternative CPAs were conducted with the supplementation of ectoine (E) and/or L-proline (P) in the freezing medium, as described in a previous study (20 mM L-proline, 500 mM ectoine, 20 mM proline + 100 mM ectoine). 24 Samples were frozen in a programmable freezer CM2000 (Carburos Metalicos, Spain). The chamber was first cooled to 1°C and held for 10 min, allowing the samples to equilibrate with the chamber temperature. The cooling rate applied was 10 K/min from 1°C to −30°C and 3 K/min from −30°C to −80°C. 28 After a 10 min equilibration step at −80°C, samples were transferred to a −150°C freezer within a box which was previously cooled to −150°C. This protocol was reported to be optimal for cryopreservation of this cell line. 24 The samples were stored at −150°C for at least 3 days before thawing.
Thawing and recultivation
Samples were thawed at 37°C in a water bath by continuously shaking for a period of 150 seconds. Cells were then transferred to a 15 mL centrifuge tube. In order to dilute Me2SO, 9 mL of pure pre-cooled DMEM (4°C) was added to each centrifuge tube drop wise over a time period of about 10 min to avoid osmotic stress. Cells were then pelletized by centrifugation and re-suspended in 1.5 mL cell culture medium. The entire procedure was performed on an ice bath.
Cell viability analysis
The membrane integrity of thawed cells was assessed using trypan blue dye exclusion, and adherence capacity was used to calculate the efficiency of recultivation. To measure cell membrane integrity directly after thawing, 0.5 mL of this cell suspension was mixed with 0.5 mL cell culture medium and measured with Vi-CELL™ cell viability analyzer. The remaining 1 mL cell suspension was divided into two parts, each of which was mixed with 2 mL cell culture medium and then cultivated for 24 h in 6-well plates.
After this recultivation period, the number of attached cells was counted with Vi-CELL™ cell viability analyzer. Cells that could adhere to the surface of the tissue culture plates were considered as viable and expected to retain proliferative abilities.
To evaluate the efficiency of cellular adherence capacity as a measure of functionality after thawing, the number of cells attached to the cell culture plate after a 24 h recultivation period was considered as output and was divided by the input number of cells which was 2.25 × 105 (one-third of cells per cryovial). The resulting ratio was defined as efficiency of recultivation. Samples without thermal treatment were used as control.
The results of membrane integrity as well as cell viability analysis were statistically analyzed using Student's t-test and one-way–ANOVA analyses with the Tukey method (using GraphPad Prism software).
RNA extraction and RT-PCR analysis
The total RNA was isolated from cultured cells using the RNeasy mini kit (Qiagen) following the manufacturer's instruction. cDNA was synthesized using 1 μg total RNA with M-MLV reverse transcriptase (Invitrogen) in a 25 μL volume. PCR conditions included initial denaturation at 94°C for 5 min, followed by 28 cycles of denaturation at 94°C for 30 sec, annealing at 60°C (GAPDH and ß-actin) and 55°C (HSP70) for 30 sec, and extension at 72°C for 30 sec,, with a 72°C extension cycle for 10 min at the end. The PCR products were analyzed by gel electrophoresis on 2% agarose and stained with ethidium bromide (10 μg/mL), following which they were visualized on a UV transluminator and photographed by a Gel Doc System™ (BioRad). GAPDH and ß-actin were used as an endogenous control in RT-PCR. Primer sequences for RT-PCR are listed in Table 1.
Western blot analysis
For total protein extraction, cells were incubated overnight at 4°C with ice-cold lysis buffer composed of 25 mM Tris–HCl (pH 7.4), 25 mM sucrose, 150 mM NaCl, 0.1% SDS, 1%Triton-X, and 1 mM EDTA. The protein concentration of each cell lysate was determined by a BCA assay (Pierce™ BCA Protein Assay Kit, Thermoscientific). The cell lysates were then mixed with SDS sample buffer composed of 100 mM Tris–HCl, 20% glycerin, 200 mM DTT, 4% SDS, 0.2% bromophenol blue, pH 6.5, heated at 95°C for 3 min, and 7.45 μg protein was loaded into each well of a 4% stacking and 12% running gel for SDS-PAGE (SDS-polyacrylamide gel electrophoresis).
After SDS-PAGE, proteins were transferred onto a nitrocellulose membrane (0.2 μm; Roth, Karlsruhe, Germany) by a semi-dry blot technique, blocked with a solution of 5% nonfat dry milk and 0.1% Tween-20 in tris-buffered saline (TBS). Primary antibodies against Hsp70 and β-actin (mouse anti-Hsp70 (ThermoScientific) and mouse anti-β-actin (Cell Signaling) were diluted using blocking solution to 1:1000 and 1:5000, respectively, and applied to the nitrocellulose membranes overnight at 4°C. The blots were then incubated with horseradish peroxidase (HRP)-conjugated goat anti-mouse secondary antibody (1:2500 in blocking solution, Santa Cruz, CA) for 1 h at 4°C. HRP signals were detected with super signal west pico chemiluminescent substrate (Thermo Scientific Pierce Protein Biology Products) and recorded with a 16 bit Kodak CCD Sensor camera (KAF 3200ME) system (Intas, Göttingen, Germany).
Results
Effects of thermal activation on cryopreservation outcome
Cells were incubated at 37°C for 4, 10, and 12 hours after heat or cold shock and before freezing with various Me2SO concentrations. These different recultivation periods were investigated to determine the amount of time required for optimal HSP expression level in order to protect cells during subsequent cryopreservation. Control samples were incubated at 37°C and cryopreserved with different Me2SO concentrations without preceding hypo- or hyperthermal treatment.
Ten hours of recultivation after heat treatment proved to have the most favorable effects on the membrane integrity (MI) of cells (Fig. 1B). This pretreatment, however, did not improve the efficiency of recultivation (ER) (Fig. 1D). A significant increase in ER was observed only after 12 hours of hyperthermal pretreatment (Fig. 1D). Also hypothermal activation showed a more considerable effect after 12 hours (Fig. 1A and 1C). This was true for all the tested Me2SO concentrations as can be seen in Table 2.
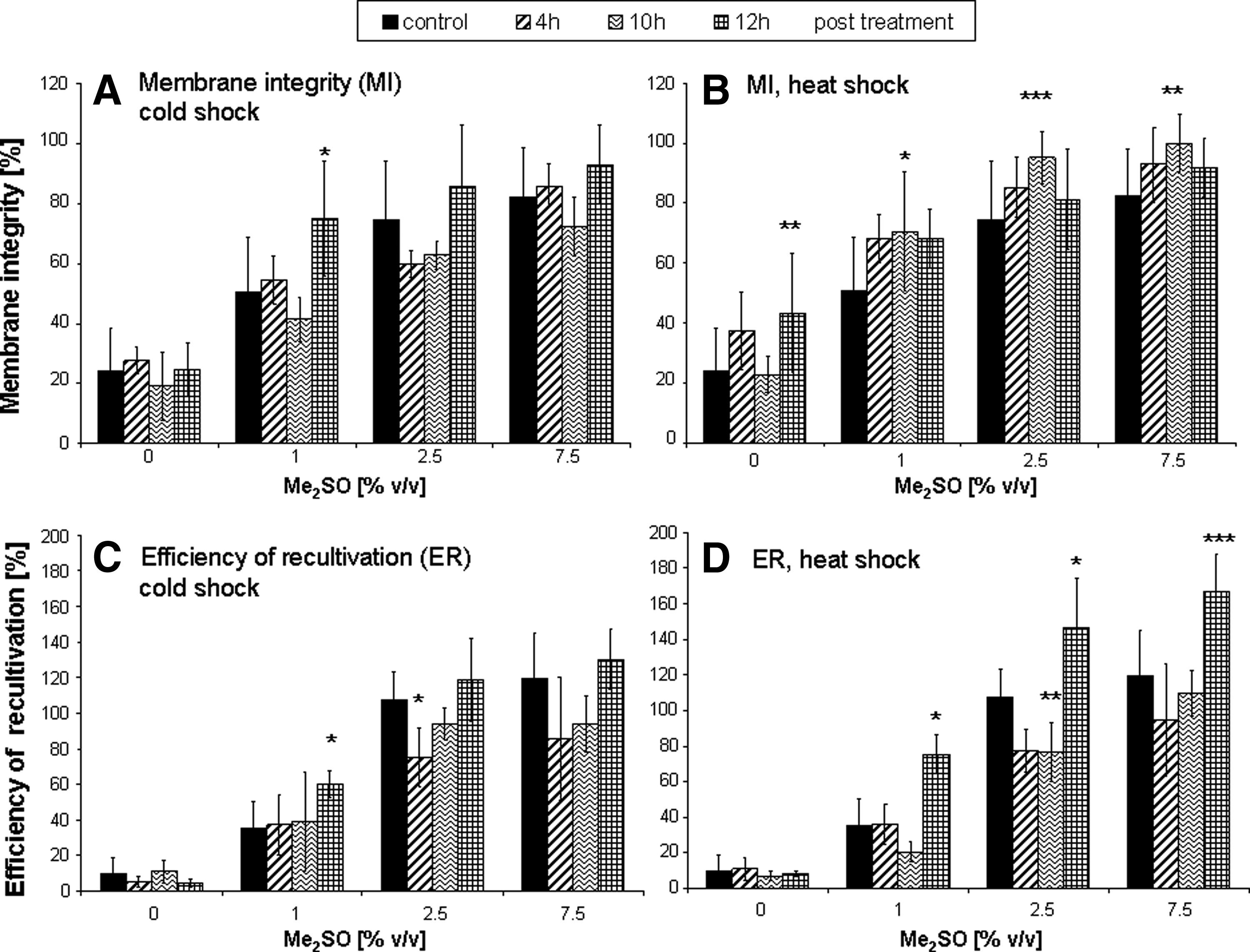
Post activation period. Membrane integrity (MI) measured directly after thawing. MI after cold treatment
Values are calculated for total sample volume, original cell count was multiplied with 3. CS, 30 min cold shock pretreatment; HS, 30 min heat shock pretreatment, Mean values ± SEM.
PCR analysis of differentially treated cells revealed an increased HSP70 gene expression at 4 and 12 hours of incubation after the thermal pretreatment (Fig. 2). Exposure of cells to 43°C increased the HSP70 expression even more than 45°C, which served as a positive control for heat shock stress. 29 Preincubation of the cells at 4°C led to a slightly enhanced induction of HSP70 gene expression after 12 hours.
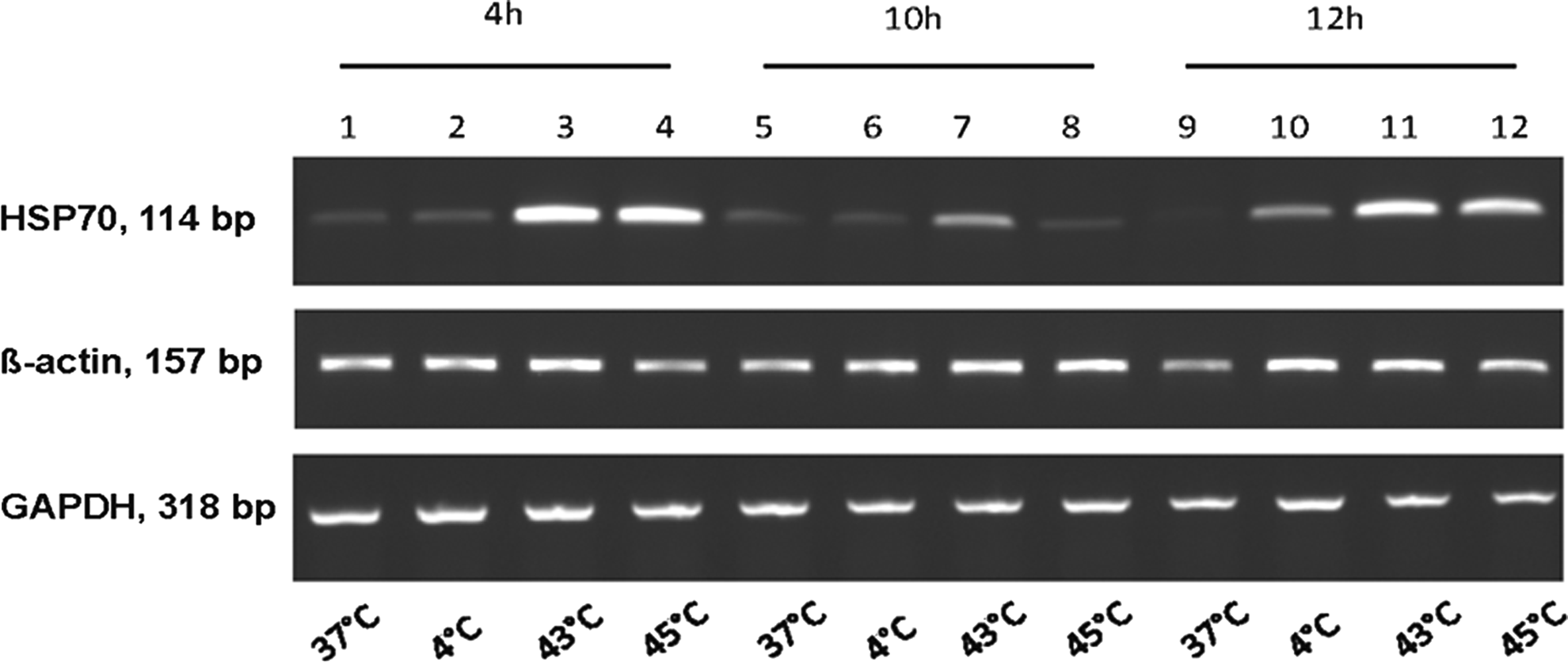
Analysis of heat-shock protein gene (HSP70) expression by RT-PCR after 4, 10, and 12 h incubation post hypo- or hyperthermal treatment. HSP70 expression level from untreated cells (37°C) is considered as base-level expression. Expression level of glyceralaldehyde 3-phosphate dehydrogenase (GAPDH) and ß-actin served as internal controls.
These results were corroborated by Western blotting analyses, where the highest heat shock protein expression was detected after activation by heat (43°C and 45°C). The HSP70 protein expression level appeared to be highest when cells were cultured for 12 hours after heat shock treatment (Fig. 3). All further experiments were therefore performed with a post activation period of 12 h.
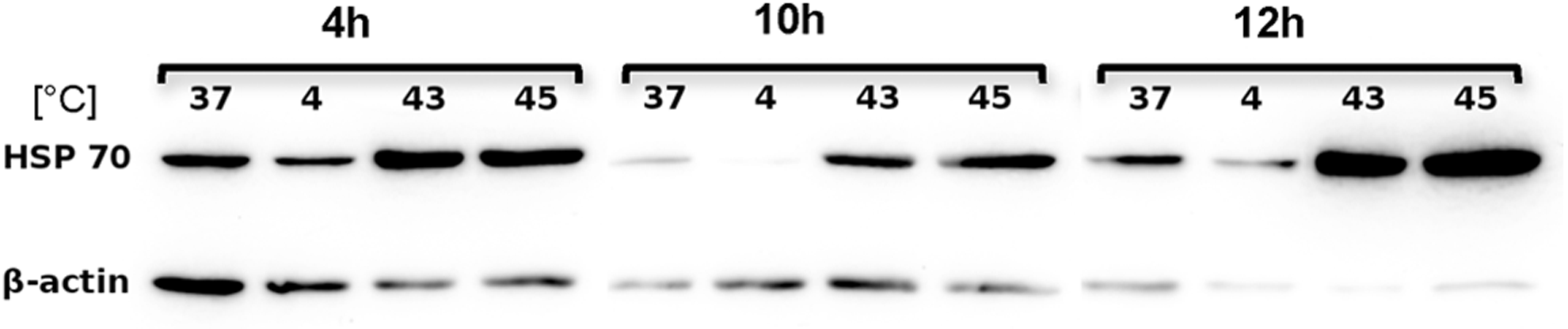
Western blot analysis of heat-shock protein 70 (HSP70) expression after 4, 10, and 12 h recultivation post hypo- or hyperthermal treatment. HSP70 expression level from untreated cells (37°C) was considered as base-level expression. Expression level of ß-actin served as an internal control.
Thermal activation and compatible solutes
Since thermal activation proved to enhance cell survival more than Me2SO alone, it was then tested if supplementation with additional compounds would allow further reduction of the Me2SO concentration used for cryopreservation. In a previous study, 24 supplementation of freezing media with L-proline and ectoine had favorable effects on post-thaw cell viability, as assessed by the efficiency of recultivation. Therefore these compatible solutes were investigated in combination with hyper- and hypothermal treatment and variable Me2SO concentrations, the results of which are shown in Table 3 and Figure 4.
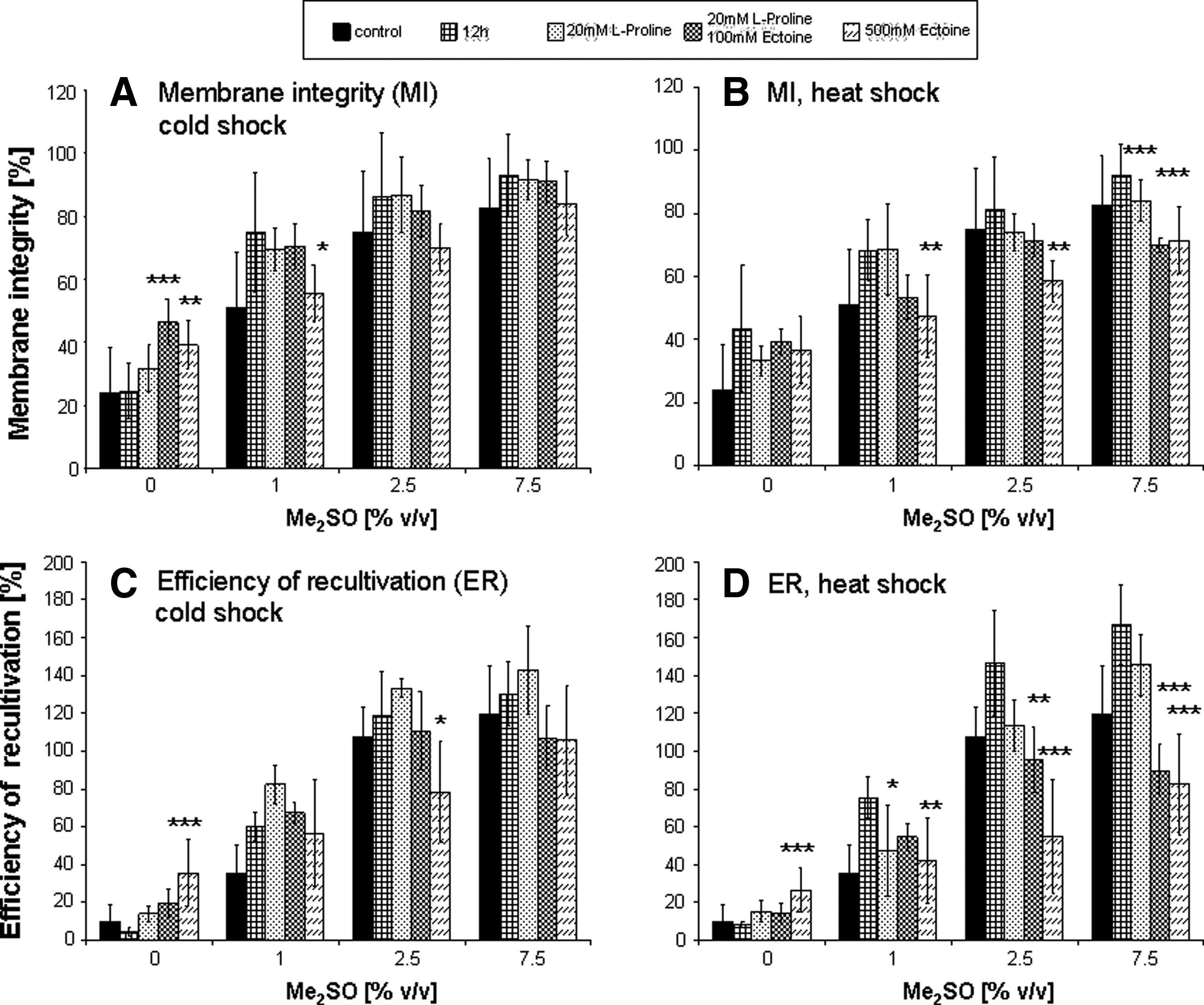
Effect of thermal activation in combination with compatible solutes as additional CPA. Membrane integrity (MI) measured directly after thawing. MI after cold treatment
Values are calculated for total sample volume, original cell count was multiplied with 3.
CS, 30 min cold shock pretreatment; E, Ectoine; HS, 30 min heat shock pretreatment; P, L-proline; Mean values ± SEM.
Cell structure (as measured by membrane integrity, Fig. 4A and 4B) was found to be supported by both heat and cold preconditioning only in the presence of lower Me2SO concentrations (0 and 1%). Supplementation with these compatible solutes appeared to have no additional effects when compared to the thermal activated control with 12 hour post-cultivation period alone.
This observation was supported by the results of the efficiency of recultivation (Fig. 4C and 4D). Hypothermal activation combined with the addition of 20 mM proline had considerably favorable effects independent of Me2SO. Cryopreservation with 500 mM ectoine revealed a substantial improvement in the number of adherent cells after thawing, as compared to the cells frozen without Me2SO. This positive effect was not observed with addition of 2.5 and 7.5% Me2SO to the freezing medium. Cells incubated at 37°C and cryopreserved with different Me2SO concentrations without preceded thermal treatment served as one control. Additionally control samples were heat shocked and cryopreserved after 12 hours post-activation period.
Discussion
The expression of heat shock proteins is an effective cellular protection mechanism in response to adverse physiological and environmental conditions, and applications based on the beneficial effects of heat shock preconditioning have been previously investigated.30–32 This mechanism can be used to mitigate the impact of severe conditions, 33 which also affect cells during freezing and thawing and to ensure the preservation of structural as well as functional integrity of cryopreserved cells. This is particularly interesting as the currently used cryopreservation protocols worldwide routinely contain Me2SO, which is widely recognized as being toxic to cells in higher concentrations.34,35 The reduction or replacement of Me2SO as a cryoprotective agent is thus a crucial step towards the production of safe cryopreservation techniques for medical purposes.
In the present study, the production of heat shock protein 70 as a known intrinsic cellular stress protection mechanism was induced by thermal activation. After incubating the HPMEC ST-1.6R cells for a period of 30 min at 43°C and 4°C respectively, the expression of HSP70 was studied. Different post activation periods were investigated to determine the time required for an optimal HSP70 expression of these cells prior to cryopreservation. The results show that, for heat activation, although the membrane integrity of cells was retained in the most favorable manner after 10 h, the functional integrity (as measured by cell adherence, ER) was best preserved by a 12 h recultivation period after the thermal activation. Compared to untreated controls, it was possible to increase the efficiency of recultivation (ER) by more than 45% by thermal activation. By this pretreatment it was also possible to reduce the Me2SO concentration to 2.5% (v/v) and achieve a 20% improved ER compared to the respective controls.
In contrast, a hypothermal pretreatment was not as effective as hyperthermal activation. Both structurally (MI) and functionally (ER), at the most a 10% increase could be observed. Possibly an increase in the ambient temperature represents increased stress for the endothelial cells as compared to temperature reduction. The production of HSP is induced more strongly by heat shock than by cold stress, which may be a measure of the level of stress experienced by cells.36–38 Our results show that recultivation period of 12 hours was required in order to achieve the most favorable cryopreservation outcome as assessed by cell viability analysis. This observation is consistent with the results of S. Wang et al. 39 who found the highest HSP production between 3 and 12 hours after treatment, as well as with the findings of Burgher et al., 16 although they wanted to use these effects to increase the efficiency of cryosurgical treatment. In contrast to P. Wang et al. 18 who also describe an increased freezing tolerance in temperature pretreated HeLa cells, Park et al. 17 did not find a protective effect in normal human fibroblasts. As hibernating insects are known to respond to cooler environmental temperatures by HSP production, 13 freeze tolerance may be induced by other cellular pathways in mammalian endothelial cells.
It was shown in previous studies24,40 that the reduction in the Me2SO concentration for cryopreservation could be achieved by the supplementation of freezing media with compatible solutes (CS) such as ectoine and L-proline. A reduction in the Me2SO concentration to 5% was possible without compromising the cellular adherence capacity. This finding was adapted in the current study and coupled with the activation of heat shock protein induction by temperature alterations.
Our results, however, indicated that heat activation alone was sufficient to enhance post-thaw cellular adherence and that the addition of CS had no additional observable positive effects on cell viability. Only after exposure to lower temperatures, a minor increase in the ER was detected as a result of lower stress-induced cell protection. Our studies have shown that hyperthermal treatment followed by 12 hours of post activation period leads to improved membrane integrity and adherence capacity of HPMEC cells after cryopreservation. By this treatment, a reduction of the cytotoxic CPA Me2SO to up to 2.5% was possible. Increased HSP70 expression in hyperthermally activated cells after 12 hours as depicted by RT-PCR and Western blot analysis showed the highest survival in these cells, suggesting that upregulation of HSP70, as assessed by hyperthermal treatment, seem to have a protective function against cryopreservation stress.
However, the upregulation of HSP70 may not be the only factor involved in heat shock-induced cell protection and other HSPs and their roles need to be studied further. Overexpression of HSP70 or knocking down its expression through RNAi without thermal treatment can further elucidate the role of HSP70 in cryopreservation of HPMEC ST-1.6R cells. Moreover, it should also be mentioned that the expression of HSPs varies across different cell types, as well as the cryopreservation protocol employed. Furthermore, viability analysis alone does not provide information on the more subtle cellular processes that may be altered during cryopreservation.
Therefore a more thorough investigation of cell proliferation over a prolonged time period should be examined in parallel with other gene expression studies. Heat shock protein induction, in combination with other stress inducers, may serve as targets that stimulate endogenous protective mechanisms in response to stressful conditions such as cryopreservation. 41 In future work, therefore, an induction will be achieved by different nonstressing stimulants. This should lead to a protection strategy that allows long-term storage of three-dimensional tissues in the future.
Footnotes
Acknowledgments
This work was supported by funding from the Deutsche Forschungsgemeinschaft (German Research Foundation) for the cluster of excellence REBIRTH (From Regenerative Biology to Reconstructive Therapy – EXC 62/1). We are grateful to the Institute of Biophysics in the Leibniz Universitaet Hannover for allowing us to use their CCD camera system for recording our Western blotting results.
Author Disclosure Statement
There are no conflicting financial interests.