
Abstract
Context:
Biobanks of frozen human normal and malignant tissues represent a valuable source for “omics” analysis in translational cancer research and molecular pathology. However, the success of molecular and cellular analysis strongly relies on the collection, handling, storage procedures, and quality control of fresh human tissue samples.
Objective:
We tested whether under vacuum storage (UVS) effectively preserves tissues during the time between surgery and storage for “omics” analyses.
Design:
Normal and matched tumor specimens, obtained from 16 breast, colon, or lung cancer patients and 5 independent mesenchymal tumors, were dissected within 20 minutes from surgical excision and divided in three to five aliquots; for each tissue sample, one aliquot was snap-frozen in liquid nitrogen (defined as baseline or T0 samples), and the other portions were sealed into plastic bags and kept at 4°C for 1, 24, 48, or 72 hours under vacuum and then frozen. The tissue and molecular preservation under vacuum was evaluated over time in terms of histomorphology, transcription (Illumina microarrays), protein (surface-enhanced laser desorption/ionization-time of flight/mass spectrometry and Western blot), and metabolic profile (nuclear magnetic resonance spectroscopy).
Results:
Tissue morphology, Mib-1, and vimentin immunostaining were preserved over time without signs of tissue degradation. Principal variance component analysis showed that time of storage had a minimal effect on gene expression or the proteome, but affected the preservation of some metabolites to a greater extent. UVS did not impact the RNA and protein integrity or specific phosphorylation sites on mTOR and STAT3. Measurement of metabolites revealed pronounced changes after 1 hour of storage.
Conclusions:
Our results show that UVS can preserve tissue specimens for histological, transcriptomic, and proteomic examinations up to 48 hours and possibly longer, whereas it has limitations for metabolomic applications.
Introduction
R
On this basis it could be expected that comprehensive genomic, proteomic, and even metabolic profiling representative of the biological complexities of health and disease might replace single diagnostic biomarkers. In fact, with the introduction of new genomic technologies, such as tissue-based RNA microarrays, patterns of gene expression able to stratify tumors according to their molecular features and predict clinical outcomes have been discovered in various cancer types. 5
Thus, storage of tissues with effective preservation of morphology, proteins, nucleic acids, and metabolites for research and diagnostic purposes is the main goal of human tissue biobanks. 6 As a consequence, collecting surgical specimens that can be used for these analyses has become a mandatory issue for assessing the correlation between clinical features and molecular data, in the majority of current clinical trials. 7 However, variability in tissue handling and processing of surgical specimens may affect the reproducibility and interpretation of results. Several preanalytical variables (including tissue manipulation, warm or cold ischemia, and storage conditions) can potentially alter apparent expression levels and adversely affect the reliability of translational studies. Thus, markers of the sample's quality are needed to monitor tissue and molecular preservation.
Because RNA is considered the most degradable molecule in unfixed tissues, preservation of RNA integrity was proposed as a general quality indicator in fresh frozen tissue biobanks. Indeed, “warm ex-vivo ischemia” during surgery (from blood vessel ligation to surgical excision time), “cold ischemia” (from excision to freezing), preservation treatment of the tissue, type and length of storage, and specimen type are just some of the several factors affecting RNA quality and stability. Furthermore, it has been shown that warm ischemia during surgery may result in significant gene expression changes,8,9 and results based on the analysis of partially degraded RNA may be unreliable and should be interpreted with caution.
The examination of protein biomarkers, particularly of phosphoproteins, is another critical step in the assessment of tissue quality. 10 Since preanalytical factors, such as ischemic duration, can have a significant impact on the ability to detect active signaling pathways in tissues, 11 phosphoprotein markers of cell signaling have been widely studied in both translational research and clinical settings, making their determination crucial for effective treatment decisions. Metabolomics is the upcoming new branch in the “omics” sciences. Despite several intrinsic limitations that still remain, currently metabolomics research in cancer tissues is being used to (i) better understand its complex heterogeneous nature, (ii) discover novel metabolic diagnostic cancer biomarkers in the clinic and monitor them during therapeutic intervention, and (iii) discover pathways involved in cancer that could be used for new targets.12–14
To preserve tissues during the time between surgery and sample processing and storage, an alternative procedure to formalin fixation has been proposed: under vacuum storage (UVS) by sealing of tissues in plastic bags at 4°C until processing. Safety and advantages linked to this UVS procedure have already been reported elsewhere.15,16 Moreover, the UVS approach seems to be convenient also in financial terms as it could diminish the usage and disposal of hazardous chemicals such as formalin. 17 Hence, we decided to evaluate, for the first time, the effect of the time-to-freezing, at various “omics” levels, on tumor samples preserved in UVS conditions, with the aim of identifying for each approach the time window within which the tissue could be considered sufficiently well preserved to enable informative analyses.
Materials and Methods
Ethics statement
All patients whose biological samples were included in the study acknowledged informed consent to donate for research purposes tissue specimens left over after completing diagnostic procedures. The Independent Ethics Committee of Fondazione IRCCS Istituto Nazionale dei Tumori of Milan (INT-MI) approved the use of these samples for this specific study in the framework of a project in biobanking quality control.
Study design and sample collection
To avoid or limit artifacts arising from technical aspects, all the technical procedures for sample collections and processing were standardized and each technical procedure was performed by the same operator in the same processing facility.
The tumor and normal matched samples analyzed in the experiments were prospectively collected from 21 patients who underwent surgical resection at the INT-MI and whose tumors were representative of the various pathologic stages of each tumor type (Table 1). Paired tumor–normal samples were obtained from eight breast, five colon, and three lung cancer patients, and tumor tissue only was collected from five mesenchymal tumor patients. Within 20 minutes from surgical excision, neoplastic samples were cleaned of necrotic or fat tissue and specimens were dissected and divided in three to five aliquots. For each tissue sample, one aliquot was snap-frozen in liquid nitrogen (defined as baseline or T0 samples) and the other portions were sealed into plastic bags labeled with identification codes and kept at 4°C for 1 (T1), 24 (T2), 48 (T3), or 72 (T4) hours in the vacuum apparatus (Tissue-safe®; Milestone, Bergamo, Italy) before being snap-frozen in liquid nitrogen and then stored at −80°C.
DD, dedifferentiated; NA, not available; WD, well differentiated.
Analyses of the preservation of histomorphology, the peptidome, and Western blot experiments were performed on all samples (21 patients, 163 specimens). In the case of gene expression (15 patients, 84 specimens) and metabolic profiling (17 patients, 132 specimens), not all samples were suitable for analysis, due to the low quantity or poor quality of recovered material. A total of 66 specimens were processed for all techniques (Supplementary Table S1; Supplementary Data are available online at www.liebertpub.com/bio).
Tissue morphology and immunohistochemistry
Sections of 3 μm were stained with hematoxylin and eosin (H&E) according to routine procedures and evaluated microscopically for tissue morphological preservation. Immunohistochemical detection was performed to evaluate the preservation and integrity of specific epitopes, using an automated slide-processing platform (Ventana BenchMark AutoStainer; Ventana Medical Systems, Tucson, AZ) and the following primary antibodies: anti-Ki67 monoclonal antibody (clone MIB1, diluted 1:100; Dako, Glostrup, Denmark) and anti-vimentin monoclonal antibody (diluted 1:400; Dako). Positive and negative controls (omission of the primary antibody and IgG-matched serum) were included for each immunohistochemical run.
Gene expression profiling
In the case of matched tumor–normal samples (breast, colon, and lung), only patients with paired samples available at all time points were processed. Lung samples were not included in gene expression analysis because paired material was not available for all time points after accounting for quality controls. Due to this selection, 90 specimens were processed (Supplementary Table S1).
After UVS, all tissue samples were stored at −80°C until RNA extraction. Total RNA was extracted from 10 to 20 mg of tumor samples and from 30 to 40 mg of normal samples. Tissues were mechanically disrupted and simultaneously homogenized in the presence of QIAzol Lysis reagent (Qiagen, Valencia, CA), using a Mikro-dismembrator (B. Braun Biotech International, Melsungen, Germany). RNA was extracted using the miRNeasy Mini kit (Qiagen) according to the manufacturer's instructions. Purification and DNase digestion were performed using two different kits: RNeasy MinElute Cleanup (Qiagen) was used for up to 45 μg of RNA, while RNeasy Mini kit (Qiagen) was used for RNA ranging between 45 and 100 μg. RNA concentrations were measured with the NanoDrop ND-100 Spectrophotometer (NanoDrop Technologies, Wilmington, DE), and RNA quality was assessed with the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA) using the RNA 6000 Nano kit (Agilent Technologies). The RNA Integrity Number (RIN) 18 was determined using the software provided by the manufacturer.
Briefly, 800 ng of total RNA was reverse transcribed, labeled with biotin, and amplified overnight using the Illumina RNA TotalPrep Amplification Kit (Ambion; Life Technologies, Grand Island, NY) according to the manufacturer's protocol. One microgram of the biotinylated cRNA sample was mixed with the Hyb E1 hybridization buffer containing 37.5% (w/w) formamide and then hybridized to array HumanHT-12-v3 expression BeadChip (Illumina, Inc., San Diego, CA) at 58°C overnight (18 hours). Array chips were washed with the manufacturer's E1BC solution, stained with 1 μg/mL Cy3-streptavidin (Amersham Biosciences; GE Healthcare, Piscataway, NJ), and scanned with the Illumina BeadArray™ Reader.
Raw expression data for a total of 90 samples were collected from scanned images using Illumina BeadStudio v3.3.8 (Illumina, Inc.) and processed using the lumi package 19 of the Bioconductor project version 2.13. Probes from gene expression data were reannotated using the illuminaHumanv4.db package. Quality control was performed on raw and processed data using a combination of four criteria: detection rate, distance to centroid, average correlation with other samples, and boxplot distribution. Samples failing more than one parameter were identified as outliers and removed. After quality control, two samples from colon cancer patients at 72 hours (T4) were discarded; thus, for this pathology, the T4 time point was not considered in further statistical analyses.
Raw data were log2-transformed, normalized with Robust Spline Normalization, and filtered, keeping only the probes with a detection p < 0.01 in at least one sample. The detection p-value represents the confidence that a given transcript is expressed above the background defined by negative control probes. For probes mapping on the same gene, the probe with the highest detection rate (number of samples in which the probe has a detection p < 0.01) was chosen to represent each gene. In the case of equal detection rates, the most variant probe (according to interquartile range) was selected.
Protein extraction and immunoblot analysis
Tumor specimens were pulverized in a Mikro-Dismembrator II (B. Braun Biotech International). The pulverized tissue samples were recovered in an ice-cold buffer containing 50 mM HEPES (pH 7.6), 150 mM NaCl, 10% glycerol, 1% Triton X-100, 1.5 mM MgCl2, 1 mM EGTA, 10 mM Na4P2O7, 100 mM NaF, 1 mM PMSF, 1 mM sodium orthovanadate, and Complete Mini protease inhibitors cocktail (Roche, Milan, Italy) according to the manufacturer's instructions. After 30 minutes incubation with gentle rocking at 4°C, lysates were cleared by centrifugation for 20 minutes at 13,000 rpm. Supernatants were collected, and protein quantification was performed with the BCA™ Protein Assay Kit (Thermo Scientific, Milan, Italy) according to the manufacturer's instructions.
All electrophoresis (sodium dodecyl sulfate–polyacrylamide gel electrophoresis) and electroblotting experiments were performed with precast polyacrylamide NuPAGE NOVEX gels (Invitrogen, Milan, Italy) and with Hybond-C super nitrocellulose membrane (Amersham Biosciences, Little Chalfont, United Kingdom).
Proteins were transferred to nitrocellulose membranes and probed with the appropriate antibodies. Immunoreactive bands were visualized using horseradish peroxidase-linked anti-mouse, anti-rabbit, or anti-goat antiserum and detected using an enhanced chemiluminescence system (Bio-Rad Laboratories, Milan, Italy). Antibodies used were as follows: anti-phospho-STAT3 (Ser727), anti-phospho-AKT (Ser473), anti-phospho-AKT (T308), anti-phospho-mTOR (Ser2448), and anti-mTOR from Cell Signaling Technology (Danvers, MA); anti-STAT3 from Millipore (Billerica, MA); anti-AKT from BD Transduction Laboratories (San Jose, CA); and anti-Tubulin β and anti-α-SMA from Sigma (St. Louis, MO).
For two-dimensional gel electrophoresis (2-DGE), 130 μg of tissue extracts was used as previously described by Salvi et al. 20 The gels were silver stained.
Samples were processed using IMAC30 surface biochips (Bio-Rad Laboratories, Hercules, CA), as described previously, 21 and 30 μg was applied to an IMAC30-Cu2+ surface. ProteinChip arrays were read using a ProteinChip Reader (PCS 4000 Enterprise; Bio-Rad Laboratories, Hercules, CA) with ProteinChip Software (version 3.0.7; Bio-Rad Laboratories, Hercules, CA) according to the manufacturer's instructions.
Surface-enhanced laser desorption/ionization-time of flight/mass spectrometry (SELDI-TOF/MS) spectra were generated by averaging 530 laser shots with the parameters indicated below. All samples were tested in duplicate and analyzed as a whole. Protein extracts from more than 100 sections were analyzed by a mass spectrometer for plastic leakage, and no significant polymer contamination has been detected. Expression Differences Mapping analysis (Ciphergen Express data management software version 3.0.7; Bio-Rad Laboratories, Hercules, CA) was applied to generate a cluster peak list, which describes how a singular peak is expressed among all specimen spectra.
Metabolomic analyses
Tissues were suspended in 0.5 mL of ice-cold twice-distilled water. Aqueous extracts (from 80 to 160 mg/sample) were prepared in EtOH:H2O (70:30, v/v) according to an established protocol previously described.22,23 Samples were subjected to ultrasonication at 20 kHz with an exponential probe, 8 μm peak-to-peak, by an MSE ultrasonic disintegrator Mk2 (Crawley, Sussex, United Kingdom) and centrifuged at 14,000 × g for 30 minutes. Supernatants were lyophilized in an RVT 4104 Savant lyophilizer (Mildford, ME), and the residue resuspended in 0.7 mL D2O (Sigma-Aldrich, Milan, Italy) containing 0.1 mM 3-(trimethylsilyl)-propionic-2,2,3,3-d4 acid sodium salt (TSP) as the internal standard (Merck & Co., Montreal, Canada).
High-resolution nuclear magnetic resonance (NMR) experiments (25°C) were performed at 400 MHz (Bruker AVANCE spectrometer, Karlsruhe, Germany). Proton magnetic resonance spectroscopy ( 1 H-MRS) spectra of tissue extracts were obtained using acquisition pulses, water presaturation, data processing and data analysis were as already described. Quantification of individual metabolites was obtained from peak areas, using correction factors determined by experiments at the equilibrium of magnetization (90° pulses, 30.00 seconds interpulse delay). Additional Spectra were acquired using a T2-filtered Carr–Purcell–Meiboom–Gill (CPMG) sequence (with water presaturation) to reduce signals from the short T2 relaxation time broad resonances from lipids and macromolecules, using 2.5 seconds of water suppression before a 90° excitation pulse.
Statistical analyses
Principal variance component analysis (PVCA) 24 was used to assign the percentage of variability to known biological and technical factors present in gene expression and metabolomic and proteomic data sets. This is a hybrid method that makes use of two established approaches: principal component analysis (PCA) and variance component analysis (VCA).
Time course analysis was performed on RIN and gene expression data using a mixed-effects linear model with Time and Tissue Type as fixed variables and Patient as random. 25 All p-values were adjusted using the Benjamini–Hochberg false discovery rate (FDR). For genes with an overall FDR <0.2, a mixed-effects linear model using Time as a fixed variable and Patient as random was performed to identify tissue-specific alterations. For PAM50 classification 26 of the gene expression profiles of breast cancer patients, the Pearson's correlation coefficient between gene expression profiles and the PAM50 centroids obtained from the genefu package 27 was calculated. Samples were assigned to the subtype showing the highest correlation.
For the proteomic and metabolomic data set, we applied the same statistical approach for each tissue type separately due to the different features detected in each tissue.
Results
Histomorphological evaluation
Randomly selected tissues stored by UVS were assessed for tissue morphology with standard H&E staining and for epitope integrity with immunohistochemical staining (IHC) for tissue-specific antigens. Tissue samples after UVS at 4°C from T0 to T4 exhibited well-preserved cellular and tissue structure, with maintenance of histological zonation and uniformity, as well as a defined intensity and distribution of cellular epitope-specific staining, with minimal or absent background noise. No signs of necrosis or of tissue alteration were observed at any time point. The morphology and IHC evaluation at all time points for all samples revealed well-preserved structural integrity and a high degree of protein stability upon staining with anti-ki67 (Mib-1) and anti-vimentin antibodies (one representative sample for each type of tumor tissue at T0 and T4 in Fig. 1).
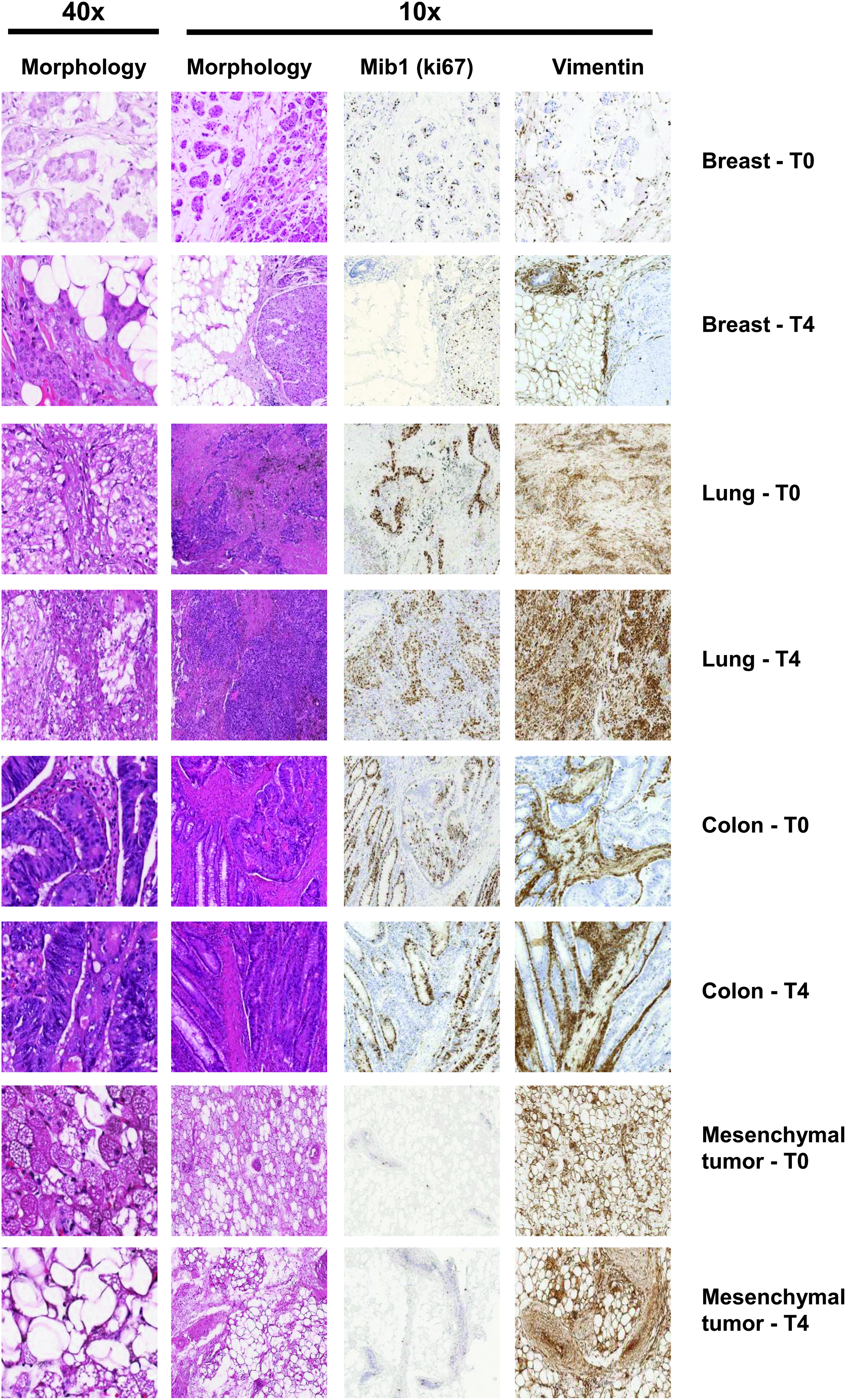
Morphologic integrity and immunohistochemical staining quality in tumor samples. Morphology (hematoxylin and eosin staining) and immunohistochemistry staining of a representative sample for each tumor type stored under vacuum for 20 minutes (T0) and for 72 hours (T4) before freezing. Immunohistochemistry staining was performed using anti-human ki67 (Mib-1) and vimentin antibodies. For hematoxylin/eosin staining, original magnification at 10 × and 40 × is reported.
Sources of variability in omics data
To determine the contribution of known biological and technical factors to the variability in gene expression and proteomic and metabolomic data, we applied PVCA. We included the time of storage, the tissue type (normal or tumor, with the exclusion of mesenchymal tumors), and the individual as known sources of variation. Results showed that in all tumor types the time of storage contributed minimally to changes in gene expression and proteomic and metabolomic data, while most of the variability could be attributed to unknown factors (residuals) for the three “omics” in almost all of the tumor types (Fig. 2). The remaining variability was explained by differences between normal and tumor tissue and by interindividual differences, with the exception of mesenchymal tumors, for which only neoplastic samples were available. In this pathology, it is notable that the analyzed samples were very heterogeneous (Table 1), probably accounting for the interindividual variability.

Principal Variance Component Analysis. Bar plot showing the amount of variability explained by the time of UVS (Time), interindividual heterogeneity (Individual), tissue type (normal or tumor, Tissue), and other unknown factors (Residuals) for gene expression and proteomic and metabolomic data. In each tissue, the Time variable is responsible for the lowest variability. For gene expression data, lung samples were not analyzed, and for mesenchymal tumors, only tumor samples were available. UVS, under vacuum storage.
Gene expression profiling
We first assessed whether RIN, which is a measure of RNA degradation, was affected by the storage time under vacuum. A considerable variability in RIN values was observed at baseline, with several samples showing a suboptimal RIN (average RIN: 5.3). However, we did not observe any significant decrease in RNA quality during under vacuum preservation (Supplementary Fig. S1). These results suggest that RNA integrity is stable for up to 48 hours of vacuum storage and probably longer as observed for breast tissues and mesenchymal tumors. The level of RNA degradation may be influenced by other factors such as the degree of tumor necrosis or tissue handling and processing.
We next performed a time course analysis to identify genes that could be altered during storage. Statistical analysis performed using all samples revealed 18 genes with a significant alteration of expression over time (FDR <20%). For these genes, we investigated more deeply their gene expression pattern in each tissue type and we found that the majority of these genes were significantly altered in expression level in normal breast and colon, while in tumor samples only a few genes were affected by storage time, especially in mesenchymal tumor samples (Fig. 3).

Heatmap reporting the average expression of differentially expressed genes (FDR <20%) at each time point in all tumor types. Significantly, tissue-specific deregulated genes are highlighted by a star. FDR, false discovery rate.
Investigating the UVS procedure in a clinical setting, we assessed whether the duration of storage could affect the reliability of a genomic predictor. Thus, we selected breast cancer patients and we assigned each individual to one of the five intrinsic breast cancer subtypes, calculating the Pearson's correlation coefficient between samples at each time point and the PAM50 centroids to verify whether the PAM50 classification was consistent throughout the storage time. Our results indicated that the classification remained constant only for samples from patient 7; in all other cases, samples from the same individual were discordantly classified, even though the misclassification occurred between subtypes with more similar PAM50 profiles, such as luminal-B and Her2-enriched or luminal-A and luminal-B (Supplementary Table S2).
Proteomic analysis
To gain insights into the capacity of UVS to preserve the proteome integrity, human malignant (n = 93) and nonmalignant (n = 70) tissue extracts were analyzed at various time points after UVS. For each sample, the protein concentration was determined to calculate the total protein yield (Supplementary Fig. S2). Storage time up to 48 hours after the UVS procedure had no negative effects on total protein yield.
Tissue extracts were then processed for Western blot to reveal changes in the expression and phosphorylation of key proteins at various time points after UVS. To establish the range of protein load in Western blot analysis, we performed the staining of the membranes (right after transfer) with Ponceau S or SYPRO Ruby (Fig. 4A). Different patterns of proteins for individual tumors and healthy control samples were observed. Several factors could account for large differences in protein content, mainly due to residual blood in differently vascularized tissues and between disease and healthy samples. Remarkably, Western blot analysis of protein extracts showed that phosphoproteins (mTOR and STAT3) were preserved up to 24–48 hours in the malignant and nonmalignant specimens that were positive for phosphoproteins (Fig. 4A). Analysis with antibodies to AKT and phospho-AKT (pS473) indicated that phosphorylation did not correlate with expression. Some differences in signal intensity were likely due to the absence of protein expression or to tissue spatial heterogeneity (especially at 0 hours for lung samples), as indicated by the variable amounts of tubulin, actin, albumin, and hemoglobin α and β detected in protein extracts (Fig. 4A and Supplementary Fig. S3). Nevertheless, in the PI3K/AKT/mTOR signaling pathway, mTOR is upstream of AKT and it phosphorylates AKT on S473. Hence, it is possible that the phosphorylated epitope could be specifically affected by ischemic conditions.
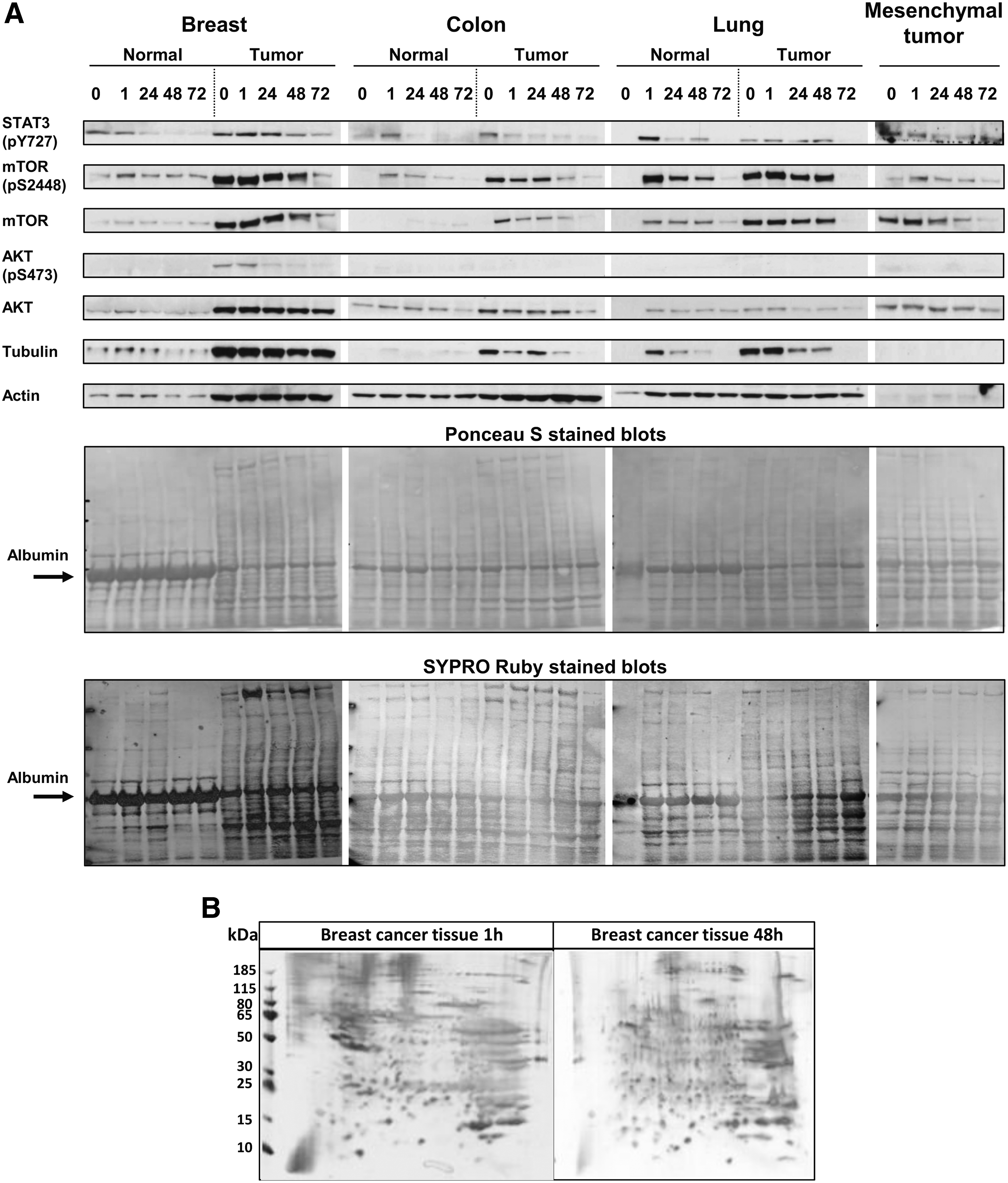
Impact of storage time on protein expression and phosphorylation.
The 2-DGE is particularly useful for screening and comparing complex mixtures such as tissue sample protein extracts. As shown in Figure 4B, the 2-DGE analysis of protein extracts (130 μg of each protein) derived from a malignant tumor tissue after 1 or 48 hours of storage, respectively, resulted in comparable spot patterns. Protein extracts yielded high-resolution spot patterns, with high numbers of protein spots showing no evidence of protein degradation.
Peptidomics studies focus only on the low-molecular weight fraction (2–30 kDa); thus, a direct analytical approach can capture the dynamic aspects of proteolytic reactions. All 163 tumor and nontumor protein extracts were processed using IMAC30 surface biochips and analyzed in a ProteinChip Reader (PSC 4000) (Supplementary Table S3). Moreover, beyond the aim of the study, data analysis showed that some peaks were differentially expressed between the cancer and normal samples. In particular, 42 out of 45 cluster peaks were significantly differentially expressed (p < 0.05) (which differentiated the nonmalignant from malignant breast tissues), 37 out of 56 had p < 0.05 in lung tissues, and 19 out of 52 had p < 0.05 in colon tissues. The ability of SELDI-TOF/MS to discriminate malignancy from nonmalignancy showed that the UVS process preserved the diagnostic peptide features.
Metabolomic analyses
To investigate the impact of vacuum preservation on metabolic profiles, 1 H-MRS analyses were performed on aqueous extracts of a number of tissue types collected at various times, and the stability of the metabolome was evaluated by integrating the metabolomic and target metabolic analyses results. The following metabolites were detected in 1 H-MRS spectra of clinical tissue samples: lactate (Lac, 1.33 ppm); acetate (Acet, 1.90 ppm); alanine (Ala, 1.45 ppm); valine (Val, 1.04 ppm); glutamate (Glu, 2.00 ppm); glutamine (Gln, 2.20 ppm); succinate (Succ, 2.40 ppm); total Creatine (tCr, 3.04 ppm); choline (Cho, 3.21 ppm); phosphocholine (PCho, 3.22 ppm); glycerophosphocholine (GPCho, 3.23 ppm); myo-inositol (m-ins, 4.05 ppm); taurine (Tau, 3.45 ppm), and glycine (Gly, 3.55 ppm).
The time course analysis showed recurrent and specific metabolic alterations for each tissue type. In particular, we observed that Cho was one of the most affected metabolites in normal and tumor breast tissue and in mesenchymal tumor tissue (Fig. 5). In colon and lung tissues, Cho exhibited a trend for increase over time, although this was not significant. This might be explained by the large scatter found in our data due to interpatient variability, heterogeneity of subsampling, and NMR peak deconvolution analyses. Besides Cho, the mean signal intensity of Ala and Val significantly increased in three of four tissue types (Supplementary Table S4). Metabolites such as creatine and phosphocreatine showed lower variations under the same experimental conditions.
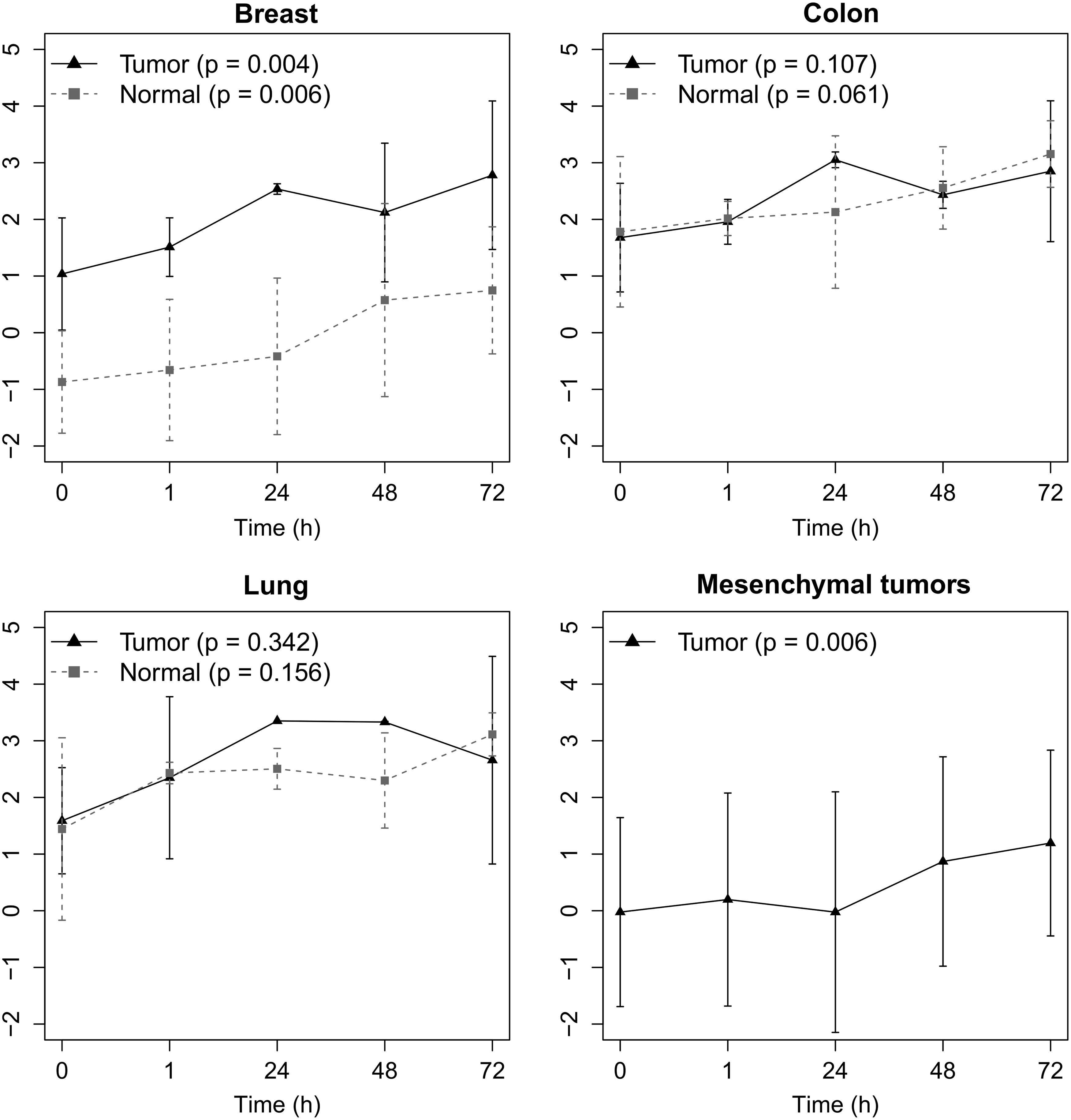
Changes of choline content in normal and/or tumor samples from breast, colon, lung, and mesenchymal tumor tissues with various times of UVS. Data are presented as mean ± standard deviation.
Discussion
Biobanks of fresh, unfixed human normal and malignant tissues represent a valuable source for “omics” analyses in translational cancer research and molecular pathology. However, the success of molecular and cellular analysis in both clinical and translational research is strongly dependent on preanalytical conditions and quality control of fresh human tissue samples. Storage of frozen tissues with intact morphology, nucleic acids, proteins, and metabolites for use in research and precision medicine is the main goal of biobanking.
In this study, with the aim of increasing the quantity and quality of biospecimens collected and to validate an appropriate procedure for multicenter studies, we tested a vacuum-based refrigerated system for tissue preservation during the time interval between surgery and banking of the samples. Bussolati et al.15,16 suggested that the vacuum sealing could preserve the tissue integrity, since the air deprivation decreased autolytic processes, thus favoring the stability of proteins and RNA. In this context, our study did not address the value of the combination of vacuum and low temperature preservation compared with that of refrigeration alone, but it was designed to evaluate the effect of UVS on histomorphology, gene expression, and protein and metabolite stability of randomly selected surgical specimens at a range of time points and from various tissue sources.
Our results show that up to 48 hours (i) tissue structure, RNA integrity, and overall epitope stability were generally well preserved, regardless of the duration of UVS, and (ii) gene expression alterations and phosphoprotein stability were not linked to degradation processes or time of preservation but to other factors, whereas metabolomic profiles were affected in a time-dependent manner.
For all samples included in this study, we observed an optimal tissue-preserving effect of vacuum sealing on morphology and immunohistochemical reactivity. These results were concordant with previous works that showed the capacity of UVS for preservation of histomorphological features.15,16,28,29
Regarding high-throughput data, we first applied PVCA to each type of “omic” data set to assess the amount of variability linked to known factors such as time of storage under vacuum and individual heterogeneity. Our results indicated that the time of storage accounted for a lower fraction of data variability compared with intersubject differences, especially for mesenchymal tumors (of which the samples belonged to a variety of histotypes). Noticeably, most variation in the data was attributed to unknown factors.
In our study, baseline samples (T0) were not subject to UVS, but were snap-frozen within 20 minutes of surgical excision. Although it was demonstrated that the time from excision to freezing can potentially alter gene expression levels and adversely affect the reliability of translational studies, 30 it has been established that the optimal time for sample collection should be within 1 hour from the commencement of the surgical procedure (within this time window the tissue remains alive and is reactive). 31 Time course analysis of RIN values, a measure of RNA integrity, showed no significant alteration over time of preservation, indicating that, despite some samples showing a suboptimal RIN value even at baseline, no pattern of further degradation was observed over 48 hours for colon samples and over 72 hours for breast tissues and mesenchymal tumors.
Analysis of gene expression profiles, however, highlighted some transcripts for which expression was significantly altered during storage time. Most of these genes coded for histone proteins or were noncoding transcripts. We would have expected to observe a decrease in the expression of genes affected by the time of storage due to the degradation of their messenger RNA (mRNA); however, all genes found to be significantly modulated were overexpressed at late time points. Interestingly, many histone transcripts have previously been reported to be affected by the time elapsed from surgery to freezing in samples left at room temperature. 9 These results indicate that the observed transcriptional alterations do not reflect patterns of degradation, but are mainly amenable to other effects, such as warm ischemia.
When we applied a genomic predictor developed for classification of breast tumors into different subtypes, we observed discordant classifications among breast tumor samples from the same patients at different UVS time points. According to our results and to previous reports, showing that expression of genes relevant for breast cancer diagnostics or classification was not affected by sample preservation or time to freezing,32,33 we hypothesize that intratumor heterogeneity or limitations intrinsic to the statistical method used to classify samples have a greater impact than time-to-freeze. However, we cannot exclude alterations in expression induced by UVS preservation due to lack of control samples preserved without UVS, and further comparisons are required.
Inhibition of kinases has become a major focus of anticancer drug development, and phosphoproteins are clinically relevant in the analysis of tumor specimens. Assessing proteome integrity is important because variation in preanalytical conditions may result in proteolysis, protein degradation, or other proteome-associated changes. Unfortunately, at present there is no comprehensive method for assessing protein quality in frozen biospecimens. 3
In terms of absolute protein yield, prolonged storage time under vacuum did not reveal any negative impact. For analytical investigation of protein degradation, we performed SELDI-TOF/MS and 2-DGE analyses of tissue extracts. These analyses indicated no significant time-dependent changes in the peptidome/degradome and displayed substantial protein integrity. The pattern of proteins seemed mostly influenced by technical variability and tissue heterogeneity.
Phosphoproteins are delicate and susceptible to both hypoxic/ischemic effects and damage from specimen handling. 34 Our results, obtained by Western blot experiments, supported the conclusion that UVS preserves phosphoproteins within tissues minimally manipulated and without depletion of the highly abundant proteins. Noticeably, phosphoproteins used in the clinical setting (such as STAT3 and mTOR) were optimally preserved up to 48 hours. In contrast, AKT phosphorylation in a number of tumor types showed a lack of reliability. Overall, signal discrepancies were independent of storage time and were probably due to tissue differences in vascularization and composition.
Pre-analytical questions are an essential and often underestimated part of clinical metabolomics study design. Using nontargeted and targeted metabolomic analyses, we reported insufficient stability after 1 hour UVS in all tissues investigated. The vacuum storage could cause an unpredictable amount of cell damage and lysis, with physical disruption of the cellular compartmentalization and changes in the molecular composition. This could be responsible for the increase in some metabolites (choline, valine). We observed a similar trend of choline accumulation in breast and ovary cancer tissue samples left on the bench at room temperature (≥2 hours) and then frozen in liquid nitrogen.23,35
Compared with macromolecules (e.g., RNA and proteins), small metabolites are probably more sensitive, in the preanalytical phase, to warm ischemia, handling, and storage. Thus, the increase in choline content could be due to ATP-independent processes (such as nonspecific lysosomal phospholipase-, phosphodiesterase-, and phosphatase-mediated tissue degradation) that occur in samples stored both under vacuum and in samples left on the bench, suggesting that this metabolite could be a useful parameter for monitoring tissue degradation in metabolomic studies.
We have to admit a limitation of our study; in fact, the small amount of available material from each specimen forced us to apply a restricted study design to avoid testing paired samples either subjected to UVS or only placed into an equivalent plastic bag at the same temperature. Thus, further studies considering this comparison are needed.
Despite some inherent limitations due to our study design, our results show that UVS is a feasible approach for preservation of tissue specimens intended for histological, peptidome, and phosphorylation analyses and that it could represent a useful tool for collecting tissue specimens from peripheral hospitals in the context of multicenter studies. This preservation method also seemed to prevent alterations in gene expression due to RNA degradation, whereas metabolites were more affected by the time of storage.
Footnotes
Acknowledgments
Financial support was received from the Italian Ministry of Health. Additional support was received by institutional funds obtained through an Italian law that allows taxpayers to allocate 0.5% share of their income tax contribution to a research institution of their choice (project Biobanking) and from the Nepente project, financed by Regione Lombardia. We thank Francesco Pastore and Gloria Morandi of Tissue Bank of Fondazione IRCCS Istituto Nazionale dei Tumori for sample collection.
Author Disclosure Statement
No conflicting financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.