
Abstract
Biobanking biological samples involve multiple handling, processing, and labeling steps. Each step may be a source of error, which if unnoticed or uncorrected may have consequences for research. We aimed to develop a simple and inexpensive genotyping method that would be valuable to detect such errors and confirm sample identity. For this purpose, seven variable number of tandem repeat (VNTR) loci were selected, analyzed by polymerase chain reaction (PCR) amplification, and organized in a PCR-based DNA profiling algorithm that proved useful to minimize the number of steps required for the procedure. Match probability calculations suggest that this method/algorithm has the potential to discriminate every participant of a biobank. As a proof of concept, the algorithm was applied on samples taken from the PROCURE Prostate Cancer Biobank. It was applied on 403 DNA samples from 101 randomly chosen patients who provided prostate tissues at surgery and blood at two to three different time points over a period of up to 7 years. A unique DNA profile requiring the analysis of no more than four VNTR loci (D16S83, D17S5, D1S80, D19S20) was successfully obtained for each of the 101 cases studied and led to the identification of two mismatches among the 403 samples evaluated (0.5% error rate). Further investigations using the same genotyping method revealed that one of the errors was due to tissue mishandling and that the other was due to tissue mislabeling. These errors, typical to the complex biobanking process, highlight the importance to implement a routine genotyping method as part of quality assurance in biobanking.
Introduction
B
Variable number of tandem repeat (VNTR) loci, or minisatellites, are chromosomal regions characterized by a single DNA sequence of 10–100 bp that is repeated back to back a variable number of times, leading to allelic variations within the human population.3,4 In forensic science, polymerase chain reaction (PCR)-based analysis of VNTRs has been exploited extensively for identity testing.5–8 However, VNTRs have gradually been replaced by short tandem repeats (STRs) and single-nucleotide polymorphisms (SNPs) mainly because multiplex assays targeting these loci are easier to develop, offer a higher throughput, and achieve a higher success rate with degraded DNA.4,9–11 In contrast, STRs, with their small repeat size of 2–6 bp, and SNPs are less suitable than VNTRs for the study of allelic variations by standard agarose gel electrophoresis, which has limited resolution, but can be performed at low cost in minimally equipped laboratories.
In this report, we describe a simple genotyping method based on the PCR amplification of seven different human VNTR loci. Optimized PCR conditions allowed for the visualization of all PCR products (i.e., alleles) by agarose gel electrophoresis and ethidium bromide (EtBr) staining. Ranking of the VNTR loci according to their power of discrimination (PD) led to the definition of a DNA profiling algorithm useful to minimize the number of PCR necessary to differentiate each biobank participant from one another. The efficacy of the method/algorithm was proven by analyzing 403 blood and tissue samples collected from 101 unrelated prostate cancer patients enrolled in the PROCURE Biobank.
Materials and Methods
The PROCURE Prostate Cancer Biobank
The PROCURE Biobank originates from the will of a patient to donate funds for the development of a biobank in view of offering to the worldwide scientific community high-quality patient samples for research. It became operational in 2007 at four Quebec University Health Centres. Essentially, 1969 patients (anonymized in the Biobank) were enrolled from 2007 to 2012 and consented to donate tissue, blood, and urine samples at prostatectomy and at follow-up visits. For each patient, the Biobank possesses several fresh frozen and formalin-fixed paraffin-embedded tissue blocks that were rapidly prepared on the day of prostatectomy. Serum, plasma, buffy coat, DNA, and total blood RNA are among the other samples that are stored at PROCURE. Overall, the Biobank contains more than 7000 samples for each category of specimens. Moreover, the bank is linked to an annotated database updated periodically that contains clinical, sociodemographic, and lifestyle data.
Sample collection and case selection
The 101 cases used in this study were randomly selected from the PROCURE Biobank cohort to fairly represent each of the four centers involved in sample collection (24–27 cases from each site). Based on a questionnaire administered to patients at enrollment, these cases were representative of the ethnic makeup of the overall PROCURE cohort (Caucasian 78.1%, African 2.5%, Asian 1.3%, Amerindian 0.7%, mixed 1.6%, no answer 15.8%).
For each case, blood samples were collected in BD Vacutainer® spray-coated K2EDTA tubes (Becton-Dickinson, Franklin Lakes, NJ) at different times, ranging from before prostatectomy to up to 7 years postsurgery. The blood was centrifuged (1180 × g, 15 minutes, 4°C), and the buffy coat fraction was removed, aliquoted, and kept at −80°C until DNA extraction. Portions of prostate tissues obtained after radical prostatectomy were frozen in optimal cutting temperature (OCT) compound, and the resulting blocks were kept at −80°C until sectioned as previously reported. 2
DNA extraction
Samples (buffy coat and tissues) were transferred from the four PROCURE centers to one site where a single individual performed all the DNA extractions and PCR. DNA was extracted from 300 μL buffy coat aliquots using the QIAamp DNA Blood Midi Kit (Qiagen, Hilden, Germany) or from cryostat sections (10 slices of 30 μm or 10–15 slices of 10 μm) of prostate tissues using the DNeasy Blood & Tissue Kit (Qiagen). For the tissues, OCT was removed before the extraction by leaving sections at room temperature for 5–10 minutes before the addition of 500 μL nuclease-free water, centrifugation, and removal of supernatant by pipetting. The DNA concentration was determined by optical density at 260 nm with a Nanodrop ND-1000 spectrophotometer (Thermo Scientific, Waltham, MA).
PCR amplification
The oligonucleotide primers used to amplify each of the seven selected VNTR loci are based on previous reports12–16 and listed in Table 1. All PCR were carried out in a MJ Mini thermal cycler (Bio-Rad Laboratories, Hercules, CA) in a volume of 25 μL containing 10–50 ng of genomic DNA, 0.15 U of KOD Hot Start DNA polymerase (Toyobo, Osaka, Japan), 1 × PCR buffer for KOD Hot Start DNA polymerase, 1.5 mM MgSO4, 0.2 mM dNTPs, 0.3 μM of each primer, and 0%–10% dimethyl sulfoxide (DMSO). VNTR-specific PCR conditions are described in Table 2.
Modified according to GenBank sequence NC_000013.11.
All reactions included an activation step (95°C, 2 minutes) before the start of the PCR cycles and a final extension step (70°C, 2 minutes) at the end of the PCR cycles.
DMSO, dimethyl sulfoxide; PCR, polymerase chain reaction.
The PCR products were separated on 0.8%–2% agarose gels ran in 1 × TBE buffer (89 mM Tris base, 89 mM boric acid, 2 mM EDTA) and stained with EtBr for ultraviolet visualization. Molecular weight markers (SimplyLoad 20 or 100 bp Ext Range DNA ladder; Lonza, Basel, Switzerland) were run simultaneously to determine the size of the amplicons from which alleles were identified. For all VNTR loci, alleles were expressed in number of repeats of the core sequence; for instance, allele “4” indicates that this allele has four repeats.
Sequencing
Some PCR products obtained after amplification of the D16S83, D19S20, and D17S24 loci were gel purified (QIAquick Gel Extraction Kit; Qiagen) for Sanger sequencing on a 3730xl DNA Analyzer (Applied Biosystems, Foster City, CA). Sequencing reactions were performed using the same PCR primers (Table 1) and were carried out at the McGill University and Génome Québec Innovation Centre.
Results
PCR amplification and allele identification
Seven VNTR loci were selected for this study: D16S83, D17S5, D1S80, D19S20, D1S111, D17S24, and RB1 (Table 2). For each locus, PCR conditions were optimized using unrelated human genomic DNA to allow clear detection of the PCR products by agarose gel and EtBr staining. Except for the D17S24 and D1S111 loci, no two loci ended up with the same optimal thermal cycling conditions (e.g., annealing temperature, number of cycles, extension time, % DMSO) rendering it impossible, in our hand, to multiplex the PCR. For all seven loci, optimized conditions led to the amplification of alleles having sizes that were in agreement with published sequences (see references in Table 3), although for confirmation, some D16S83, D19S20, and D17S24 amplicons were also directly sequenced (not shown). The difference in size between adjacent amplicons/alleles was coherent with the length of the repeated core sequence characteristic of each VNTR locus (Table 3). However, two intermediate D16S83 alleles were observed in the course of this study: one allele with a size ranging between alleles 7 and 8 (observed in one individual) and another having a size ranging between alleles 4 and 5 (observed in three individuals). Table 3 also shows, for each locus, the size of the amplicon corresponding to the most common allele observed in this study as well as the number of repeats it contains.
Small variations may exist.
Estimated based on the indicated reference.
Determination of a DNA profiling algorithm
A preliminary genotyping study was performed using the DNA extracted from the buffy coat fraction of 23 unrelated individuals. These results (Table 4) were used to rank the seven loci according to their PD and served to define a DNA profiling algorithm that has the potential to differentiate millions of people (Fig. 1). The first step of this algorithm consists in determining the D16S83/D17S5/D1S80 profile, that is, the combined profile of the three most discriminant loci. The probability that two randomly selected individuals share the same D16S83/D17S5/D1S80 profile, the match probability (MP), was calculated to be one in 1637 based on the 23 DNAs first characterized. Individuals that are indistinguishable after the first profiling step are subjected to the next steps of the algorithm (steps 2–5). These steps involve the successive analysis of the four other VNTR loci, which are evaluated one at a time until all individuals can be differentiated from one another. The calculated MP decreases significantly after each step and reaches one in more than 7 million when all seven selected loci are considered (Fig. 1).

Defined algorithm used to provide a unique DNA profile to each participant. The indicated MPs were calculated from the profiling of 23 unrelated individuals. In parentheses is the MP of each specific locus and on the right is the MP resulting from the combined analysis of multiple loci. MP was calculated as the sum of the squared frequency of each genotype at a given locus. Multiloci MP = MP1 × MP2 × MP3… MP, match probabilities.
PD = 1 − match probability.
PD, power of discrimination.
Efficiency of the method/algorithm to detect sample mispairing
To test our VNTR-based procedure, and at the same time estimate the frequency of sample mispairing in the PROCURE Prostate Cancer Biobank, the algorithm depicted above was applied to 403 DNA samples from 101 randomly chosen participants different from the 23 individuals genotyped in the preliminary study. The patients provided prostate tissues at surgery and blood at three different time points over a period of up to 7 years with the exception of one participant for whom only two blood samples were available. Out of the 101 cases analyzed, 99 showed a unique D16S83/D17S5/D1S80 profile. The DNA profiles of 10 of these cases (named 1–10) are shown in Figure 2. Analysis of the D19S20 VNTR locus (step 2 of the algorithm) sufficed to differentiate the two remaining cases (named 11 and 12, Fig. 2). The comparison of profiles between individuals was facilitated by the expression of alleles in the number of repeats associated to each amplicon (Fig. 2, right panel). With 101 unique DNA profiles, sample mispairings were easily identified and detected in 2 of the 403 samples analyzed (0.5%).
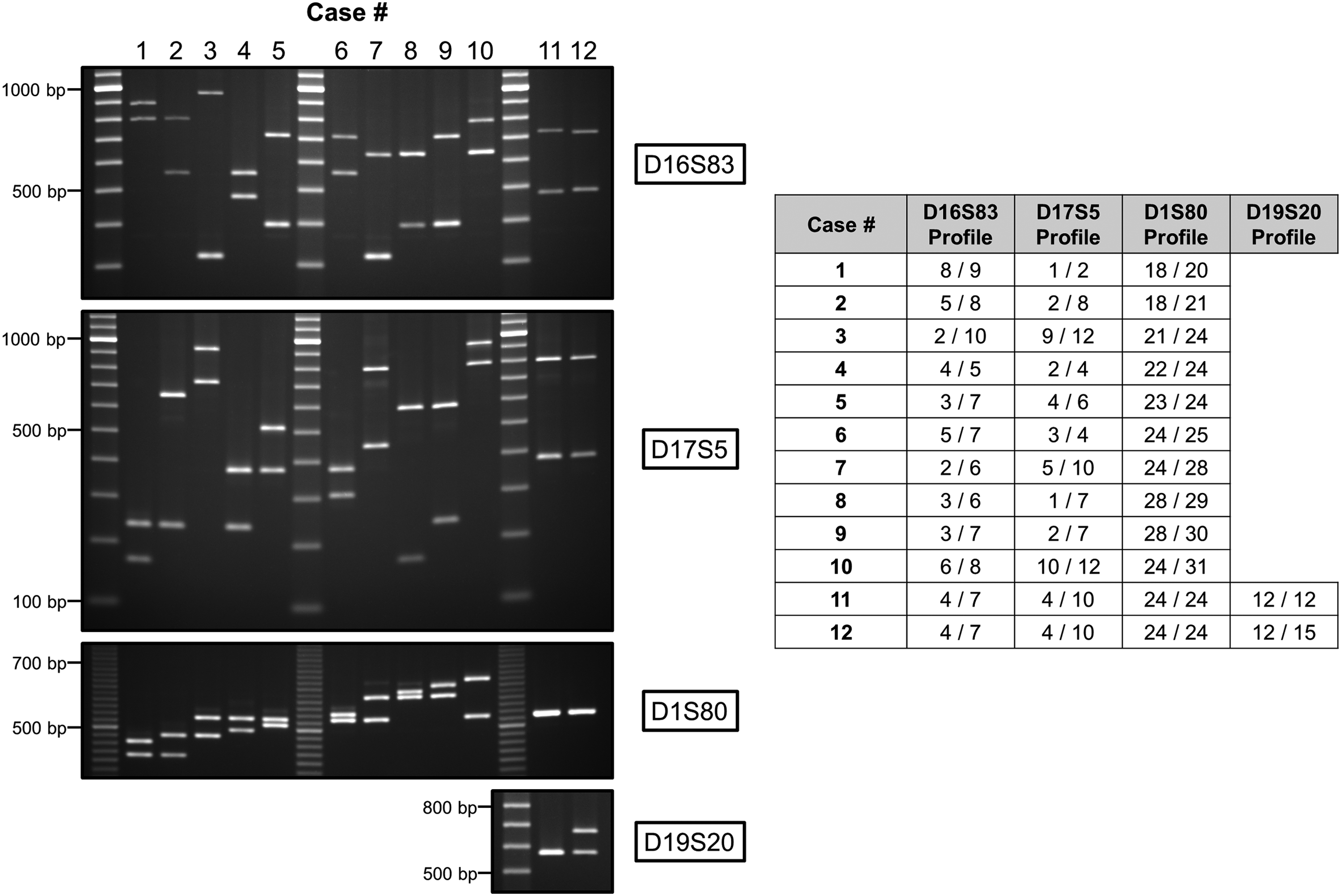
Examples of 12 unrelated cases genotyped following the DNA profiling algorithm described in this study. (Left panel) EtBr-stained agarose gels showing PCR products and alleles resulting from the amplification, in a first step, of the D16S83, D17S5, and D1S80 loci for all cases and, in a second step, of the D19S20 locus necessary to differentiate cases 11 and 12. (Right panel) DNA profiles of the same cases expressed in number of repeats associated to the alleles observed on gel; two alleles per locus per case. Molecular weight markers: SimplyLoad 20 bp (D1S80) or 100 bp (D16S83, D17S5, and D19S20) Ext Range DNA ladder (Lonza, Basel, Switzerland). EtBr, ethidium bromide; PCR, polymerase chain reaction.
Investigating mismatches
Mismatches found consisted of two tissue samples possessing a different DNA profile than the one obtained for their three different blood samples, which all had an identical DNA profile. Of note, these two tissue samples showed a DNA profile different from any other profile determined in this study, hence excluding experimental mix-ups and cross-contaminations.
To determine the underlying causes of the mismatches, further investigations were carried out and additional samples were requested and genotyped following the same experimental strategy (i.e., algorithm). One mismatch was attributed to the mishandling of a tissue block. More specifically, the wrong block of tissue was chosen from the −80°C freezer. Profiling of the DNA extracted from the correct tissue confirmed that its D16S83/D17S5/D1S80 profile matched the one already determined from this patient's blood samples (Fig. 3, left panel). The second error was due to an inversion in the labeling of tissue blocks from two patients (“A” and “B”) who underwent surgery on the same day. This inversion was noticed on that same day and indicated in the files (Biobank worksheets) of both cases, but not corrected on the samples. Consequently, the tissue block chosen for the study had the desired labeling (“A”), but belonged to the other case (“B”). The subsequent profiling of the DNA extracted from the tissue and three blood samples originally labeled as “B” confirmed the inversion that took place between these two cases (Fig. 3, right panel).
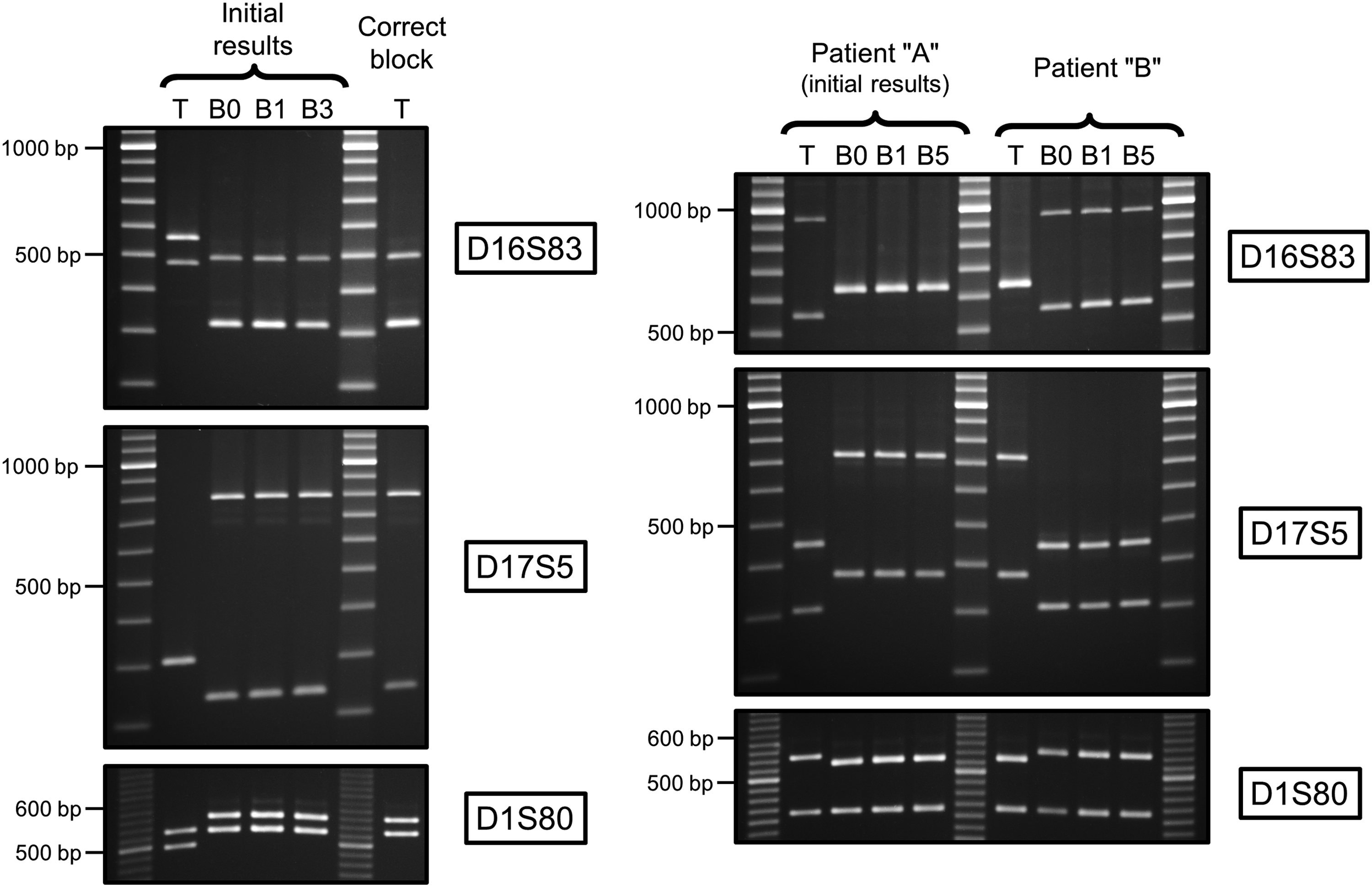
Agarose gels of the PCR products showing the D16S83/D17S5/D1S80 profiles of prostate tissues “T” and associated blood “B” samples labeled according to time of surgery, “B0,” and subsequent years (1, 3, and 5). Each series of EtBr-stained gels shows the profiles of the four samples initially analyzed and revealing for two cases mismatches between tissues and associated blood samples. The DNA profiles of additional sample(s) processed to establish the source of the error are also shown. (Left panel) Mishandling error: the wrong block of tissue was chosen from the freezer. The profile of the DNA extracted from the correct block of tissue matches the profile of the blood samples. Note the presence of a D16S83 intermediate allele (size between allele 4 and 5) observed for the four samples B0, B1, B3, and “correct” tissue (all from the same individual); compare lower band (allele 4) of “initial” tissue and upper band (allele 4+) of other samples. (Right panel) Mislabeling error: inversion in the labeling of tissue blocks from two patients (“A” and “B”) who had surgery on the same day. Samples from patient “A” were the one initially included in the study. The profile of the DNA extracted from the tissue labeled as “A” matches the profile of the blood samples labeled as “B” and vice versa. Molecular weight markers: SimplyLoad 20 bp (D1S80) or 100 bp (D16S83 and D17S5) Ext Range DNA ladder (Lonza).
Discussion
By definition, biobanking requires the collection and manipulation of several biospecimens collected from different consented patients or donors. Depending on the type of biobank, collection may be a one-time event or may be repeated over a number of years. In the prospective PROCURE Prostate Cancer Biobank, the process starts before prostatectomy (collection of blood and urine), then with the banking of prostate tissues obtained from the operating room, and continues with the periodic recollection of blood and urine in subsequent years of follow-up. In other words, several qualified persons intervene in each of the four University Health Centres to collect and manipulate samples with labeling, processing, aliquoting, and storing being part of the biobanking process. Despite the mandatory use of standard operating procedures, errors may be introduced at any time during the entire exercise. In this study, we demonstrated that the frequency of sample mix-ups throughout this relatively complex procedure is relatively low. The error rate of 0.5% (2 out of 403 samples analyzed) is reassuring and falls within the range reported for a mother and child biobank (0.8%) 19 and a colorectal cancer tissue bank (1%). 20 Nevertheless, for research, as for clinical situations, it is of utmost importance to genotype samples to certify their identity and rule out errors.
The VNTR-based genotyping method developed in the present study clearly showed its discrimination and error correction capacity. Other genotyping strategies have been reported and successfully used in biobank settings.21–24 However, these methods, which have a higher throughput and are more efficient on highly degraded DNA samples, involve the study of STR, SNP, or indel polymorphisms and depend on capillary electrophoresis or mass spectrometry, two separation techniques that may not be available to every biobank, especially those in the early phase of development. In contrast, the genotyping method described in this article is simple, necessitates only basic equipment, and, by following the defined algorithm, limits the number of PCR to the minimum to differentiate individuals. Of note, VNTR loci amplification recently showed utility in a biobank case study and sufficed to assure sample identity. 25
In our hands, it was not possible to identify a unique PCR condition that was satisfactory for all seven loci evaluated. Consequently, each locus, except for two, needed to be amplified independently of the others with its own specific thermal cycling conditions. Although it would have been useful to amplify multiple loci at the same time on the same thermocycler, the total PCR cycling time for the amplification of any locus does not exceed 60 minutes. Others have succeeded, at least in part, to multiplex some of the loci described in this study. 12 However, much longer cycling times were used (about three times longer), probably due to the use of Taq instead of KOD polymerase, and a fluorescent mode of detection was utilized. Fluorescent detection, which was not an option in our study, is more sensitive than EtBr staining and may allow for the visualization of insufficiently amplified PCR products following suboptimal thermal cycling conditions, hence facilitating multiplexing. Another possible improvement of the method, although irrelevant for a prostate cancer biobank, would be the addition of a gender determination step. For example, analysis of the SRY gene, which is present only on the Y chromosome, could be included as an extra PCR step preceding the algorithm. 26
For a biobank such as the PROCURE Prostate Cancer Biobank in which the size of the prostatectomy cohort was defined at the outset and in which no new enrollment of cases takes place, it would be relatively straightforward to determine the specific genotype profile of each participant, enter it in the biobank database, and reference it in the future if a sample needs to be identified. Indeed, this will prove to be most useful when longitudinal studies on cancer progression will be performed using blood samples added during follow-up of any given patient. The combined probability of a match at all seven VNTR loci evaluated was determined to be one in more than 7 million individuals, which is far from the cohort size of ∼2000 cases in the PROCURE Biobank setting. Moreover, this probability is likely an underestimate of the real MP, since it is based on the profiling of only 23 unrelated cases (Table 4). Indeed, the discrimination powers of the D16S83, D17S5, and D1S80 loci calculated from the analysis of all individuals genotyped in the present study (126, including the 2 mismatches) are higher (0.966, 0.962, 0.943, respectively) than the ones determined after the preliminary study. Regardless, the seven loci genotyping strategy described here will most likely fulfill the needs of banks larger than the PROCURE Biobank.
In this study, we found two mispairing errors resulting from two different types of manipulations common in any biobank: handling and labeling of samples. The source of the mishandling error was attributed to the fact that both blocks of tissue, the one that was originally chosen and the one that should have been selected, have very similar 10-digit identifying numbers. Nonetheless, a closer examination would have prevented this error. Although this type of error would be unlikely to occur in robotized sample selection, other errors may occur before and after storage. In addition, the second error in our study could have been prevented, especially since it was noted years before in the accompanying worksheets of the two prostates collected on the same day. In this case, the labeling on the samples should have been changed on the day the error was noticed. Processing errors, although not observed in our study, represent another type of error that could be associated to biobanking. Indeed, mixtures of genomic profiles in DNA extracted from single blood samples were reported and attributed to errors that occur during DNA isolation. 19 Despite their obvious preventability, human errors are likely to occur in most if not all biobanks. While the error rate must be kept at a minimum, it will probably never be nil. For this reason, the implementation of a robust genotyping method as part of the quality assurance of a biobank is highly desirable since it would help correct such errors before conducting research on often precious patient samples.
In conclusion, PCR amplification of VNTR loci, in combination with a well-defined algorithm, is a simple, reliable, and economical genotyping method that proved to be useful for the identification (and correction) of errors that may occur with sample collection and processing during biobanking. Such a method may also have its utility for other applications aimed at differentiating or matching human samples. The implementation of sample identity is a must for biobanks offering more than one sample per case for research purposes. This should be part of the quality assurance offered to investigators to give them confidence and trust in the samples used for their research to ultimately draw valid conclusions from their findings.
Footnotes
Acknowledgments
The authors wish to first sincerely thank patients who kindly donated precious samples for research. The work of partner scientists, pathologists, nurses, research associates and assistants, administrative secretaries, or other members who contributed to this Québec province-wide collective biobanking effort is recognized and particularly the implication of Ms. Phuong-Nam Nguyen and M. Saro Aprikian in this quality analysis project. This work was supported by funds raised by PROCURE for the Biobank in partnership with the Cancer Research Society of Canada since 2013. The authors wish to also thank “Le Fonds de la recherche du Québec en santé” for financial support to the Biobank in 2005–2006 and Prostate Cancer Canada in 2011.
Author Disclosure Statement
No conflicting financial interests exist.