
Abstract
In the previous decade, numerous biobanks were established and have created large markets for the storage of bioactive compounds, cells, and tissues for medical and diagnostic applications. For in vivo clinical and therapeutic purposes, it is critical to use well-defined and xeno-free components during cultivation, preservation, and transplantation of biological material. Safe and efficacious storage of bioactive molecules, cells, and tissues, without the addition of undefined medium components, minimizes risks of zoonotic disease transmission and is thus an essential and desirable prerequisite for biobanks. This gives rise to a need for well-characterized and serum-free freezing media for application in cryopreservation. For this purpose, cryobiological additives such as methylcellulose, poloxamer-188, and α-tocopherol, which have previously been shown to exhibit a cytoprotective activity, have been investigated for cryoprotection on stem cells. With this strategy, the application of fetal bovine serum (FBS) could be avoided and the concentration of toxic cryoprotective agents such as dimethyl sulfoxide (DMSO) could be reduced. Our results suggest that the viability, as well as the adipogenic and osteogenic differentiation capacity of the thawed bone marrow-derived multipotent stromal stem cells, could be maintained using a freezing medium without FBS consisting of methylcellulose, poloxamer, and α-tocopherol with only 2.5% DMSO (% v/v).
Introduction
I
The main restriction is the usage of undefined medium components such as animal serum, especially fetal bovine serum (FBS), during cell culture and cryopreservation. FBS is used in cell culture and cryopreservation media in varying concentrations (5%–95%). 2 This ill-defined cocktail of proteins is required to supply the cells with necessary nutrients and growth factors. It stabilizes the membrane, adjusts the osmotic pressure, and protects the cells from free oxygen radicals. 2 The mechanism by which serum protects the cell membrane from huge shear stresses and stabilizes the plasma membrane remains elusive. 2
For safe and successful in vivo medical applications, the elimination of FBS is essential, as it can be a source of zoonotic infections. 3 Alternatives to FBS include autologous serum and human serum albumin. However, autologous serum requires a preoperative step and is expensive. 3 Therefore, cell cryopreservation and cultivation should preferably be done without animal and human serum (xeno-free cultivation and preservation).4,5 To achieve this goal, investigation is ongoing for identifying fully defined alternatives. A promising alternative to FBS is methylcellulose. 6 Methylcellulose (MC) is a high–molecular weight nonpermeating polymer used as a substitute for serum in culture medium. 6 The function of serum proteins and MC is similar in culture 7 and it has a stimulatory effect on cells in suspension. 8 Due to its low toxicity and high viscosity, MC has been used as a component in semisolid culture of bone marrow stem cells. It was also used as a component in serum-free freezing media for cryopreservation. 9
Aside from the use of ill-defined components such as FBS, the application of potentially toxic chemicals in freezing media plays an added role in limiting the long-term storage of biological materials. In this context, application of the toxic cryoprotective agent (CPA) dimethyl sulfoxide (DMSO) is being increasingly questioned. 10 Replacing or reducing DMSO concentrations in cryopreservation has been studied in previous research.6,11–14 The feasibility of freezing cells with a reduced DMSO content (7.5%, 5%, and 2.5%) in combination with different additives such as trehalose or methylcellulose showed promising results for several cell types, including human bone marrow multipotent stromal cells (BmMSCs), 15 rat, mice, and calves MSCs, 16 peripheral mononuclear cells,4,5 as well as human MSCs. 11
In addition to the classical cryoinjuries during cryopreservation, the formation of reactive oxygen species (ROS) can occur, leading to DNA damage and resulting in decreased viability after the freeze–thaw process. Antioxidants (i.e., catalases, peroxidases, superoxide dismutases, ascorbic acid, α-tocopheryl acetate, and reduced glutathione) scavenge free radicals thereby providing protection against oxidative damage reactions such as lipid peroxidation, protein oxidation, and/or DNA damage. 17 It has already been shown that the supplementation of antioxidants to the freezing medium improves the viability of cryopreserved bone marrow cells.18,19 The antioxidant α-tocopherol seems to play a pivotal role in alleviating membrane damage caused by increased ROS production during the cryopreservation of boar semen. 17 In addition, compatible solutes (ectoin, proline) can be used to reduce the oxidative stress and/or stabilize cell membranes during cryopreservation.11,20
Another interesting chemical that can serve as a potential CPA is poloxamer 188 (Pol). It is a nontoxic, nonionic surfactant with an average molecular weight of 8400 Da and is composed of EO (polyoxyethylene) and PO (polyoxypropylene) units arranged in a basic triblock structure. This compound was found to have cytoprotective functions by protecting cells against shear stress, mechanical agitation, and increasing viability. 21 Poloxamer 188 has been shown to protect Tetrahymena cells from physical and chemical stress and prolong their survival under toxic conditions. 22 In addition, poloxamer 188 has been shown to increase the proliferation rate of frozen–thawed Caco-2 cells, as it decreased the cell surface tension thereby allowing plasma membrane resealing after wounding.23,24
In an attempt to address the current challenges of cryopreservation using high DMSO concentrations and potentially high-risk serum proteins, the present study examined the effect of cryopreservation using alternative cryoprotectants such as methylcellulose, poloxamer-188, and α-tocopherol on the viability and differentiation capacity of multipotent stromal cells from the common marmoset monkey, Callithrix jacchus. The common marmoset monkey is phylogenetically closer to humans than rodents. The ease of handling and breeding of C. jacchus makes it an attractive preclinical alternative as a nonhuman primate model and thus a powerful tool for disease and transplantation modeling.25,26 To the best of our knowledge, cryopreservation using this new CPA combination comprising MC, Pol, and DMSO has not been reported until now.
Materials and Methods
Materials
All chemicals were purchased from Biochrom (Germany), unless stated otherwise. BmMSCs were initially isolated from the tibia of the common marmoset monkey (C. jacchus, cjMSCs) as described previously. 27 No animal experiments were performed in this study.
Cell culture
cjMSCs were cultured under sterile conditions using culture media (DMEM containing FBS [15%, v/v], pen/strep [1%, v/v], and ascorbic acid [50 μM, Sigma-Aldrich, Germany]). Petri dishes (TPP), 160 mm2, were used to grow the adherent cells in a humidified incubator at 37°C and 5% CO2. For subcultivation, the culture medium was removed, and cells were detached and then centrifuged (200 g for 8 minutes). The number of cells with intact cell membrane was determined with an automatic cell counter (Vi-Cell®; Beckman Coulter GmbH). The supernatant was discarded, and cell pellets were resuspended in culture medium to obtain a concentration of 106 cells/mL and transferred into the cell culture dishes.
Cryopreservation
To test the protective properties of the CPAs on BmMSCs, cells were frozen, stored for at least 24 hours, and thawed according to our previously established protocol.12,28 The differences in efficiency of recultivation (ER), metabolic activity, and differentiation capacity were assessed. The base solution (methylcellulose/poloxamer [MP]) of the freezing media consisted of 0.1% (v/v) methylcellulose (M, 4000 cP; Sigma-Aldrich) and 1% (v/v) poloxamer 188 (P; Sigma-Aldrich) in DMEM. As an antioxidative component, α-tocopherol (T, 50 μM; Sigma-Aldrich) was also used. For each condition, a cell suspension containing a total amount of 106 cells was prepared in DMEM without supplements and placed on ice. The same volume of double-concentrated CPA solutions (5% DMSO [control], MP, and methylcellulose/poloxamer/α.tocopherol (MPT), see Table 1) was added dropwise to prevent osmotic damage. Cells were then incubated for 10, 30, or 60 minutes. As a control, the cell suspension (consisting of DMEM with 5% DMSO and 22.5% [v/v] FBS) was always incubated for 10 minutes. After the incubation time, 1 mL of cell suspension was transferred into a cryovial (1.8 mL; TPP), frozen in a controlled rate freezer CM2000 (Carburos Metalicos) either with a one-step freezing protocol (1°C/min from 4°C to −80°C) or with a two-step freezing protocol (7.5°C/min from 4°C to −30°C followed by 3°C/min to −80°C), and stored at −150°C for at least 24 hours before thawing. Cryovials were thawed in a 37°C water bath for 90 seconds until small ice crystals were visible and used for further analysis. 29 Cryoprotectant was removed from thawed cells by dilution with cold culture medium, centrifugation, and resuspending in warm culture medium, and cells were counted by an automatic cell counter (Vi-Cell; Beckman Coulter GmbH).
CPA, cryoprotective agent; DMSO, dimethyl sulfoxide; FBS, fetal bovine serum; MP, methylcellulose/poloxamer; MPT, methylcellulose/poloxamer/α.tocopherol.
Recultivation efficiency and metabolic activity
For determining the metabolic activity, 10,000 cells/well were seeded in 96-well plates (TPP) and incubated at 37°C. After 24, 48, 72, or 96 hours, 15 μL of MTT dye (5 mg/mL in PBS; Sigma Aldrich) was added to each well. The plates were incubated at 37°C for 4 hours. One hundred microliters of stop solution (10% formic acid in isopropanol) was then added to each well and incubated for 1 hour. Absorbance was then measured at 570 nm using a multiwell plate reader (Multiskan Ex; Thermo Scientific).
From the remaining cell suspension, 250 μL was seeded into a six-well plate (TPP) and incubated for 24 and 48 hours. The ER was determined by removing the supernatant, washing, and incubation with trypsin for 5 minutes. The numbers of cells with intact membranes were measured with a Vi-Cell cell counter (Beckman Coulter GmbH). ER is represented by the ratio of total number of adherent cells to initial number of cells during seeding and was considered as cell viability after cryopreservation. 12
Doubling time
Cells were thawed, counted, and a cell number of 5 × 104 cells/well was seeded into each well of a 12-well plate and incubated at 37°C and 5% CO2 until analysis. For each condition, triplicates were used. The cell number was counted for five consecutive days. Each day the cells of one well were detached and counted by an automatic cell counter (Vi-Cell; Beckman Coulter GmbH). The doubling time was calculated with the following equation:
where t is the time period, Ne the cell number at the end, and Nb is the starting number of cells.
Adipogenic differentiation
104 cells/well were seeded in 24-well plates and cultivated to 80% confluency. The culture medium was replaced with the adipogenic differentiation medium containing 10% FBS (v/v), 1 μM dexamethasone, 10 μg/mL insulin, 0.5 mM 3-isobutyl-1-methylxanthin, and 0.2 mM indomethacin (all components except FBS were purchased from Sigma Aldrich). The differentiation medium was changed after 3 days, replaced with a maintenance medium (DMEM, 10% FBS, and 20 μg/mL insulin), and cultured for 2 days. The total period of cultivation for differentiation was 20 days. After differentiation, Oil Red O staining was performed to visualize adipogenic differentiation. The wells of the 24-well plates were washed briefly, followed by cell fixation using 4% paraformaldehyde (PFA). The cells were washed with water and then with 60% (v/v) isopropanol. Afterward, the cells were incubated with Oil Red O solution (Sigma Aldrich) (stock solution: 0.35% Oil Red O in 60% [v/v] isopropanol) for 1 hour followed by three washes with PBS. The Oil Red O stained in the lipid vacuoles was eluted using 200 μL isopropanol. The eluted solution was then transferred in a 96-well plate and the absorption was read at 450 nm using a multiwell plate reader (Multiskan Ex; Thermo Scientific).
Osteogenic differentiation
Osteogenic differentiation was performed as described previously. 30 Briefly, 104 cells/well were seeded in 24-well plates. After 24 hours, the culture medium was replaced with the osteogenic differentiation medium containing DMEM, 15% FBS, 0.1 μM dexamethasone, 10 mM β-glycerol 2-phosphate, 0.05 mM L-ascorbic acid 2-phosphate, and 1% ITS premix. The cells were cultured in this medium for 3 weeks with a media change twice every week. After differentiation, von Kossa and Alizarin Red S staining was performed to visualize osteogenic differentiation. The cells were washed briefly, followed by cell fixation using 4% PFA. For Alizarin Red S staining, the cells were washed with PBS, then incubated for 1 hour with 40 mM Alizarin Red S (pH 4.1) (0.5 mL/well), eluted with 10% (w/v) cetylpyridinium chloride (0.2 mL/well) for 1 hour, and quantified by measurement of absorbance at 570 nm. Von Kossa staining was performed by incubation with 5% silver nitrate solution (50 mg/mL) in the light for 30 minutes, followed by two washes with double-distilled water. The cells were then stained with 1% pyrogallol for 2–3 minutes followed by washing and further staining with 5% sodium thiosulfate solution (50 mg/mL) for 2–3 minutes, followed by two washes with double-distilled water. The cells were finally air-dried and observed under the microscope (Axiovert M1; Carl Zeiss).
RNA extraction and quantitative real-time polymerase chain reaction
To analyze gene expression of differentiation-specific genes, quantitative real-time polymerase chain reaction (qRT-PCR) was performed using the SYBR Green and 2−ΔΔCT method. Total RNA was extracted from cells using the peqGOLD MicroSpin Total RNA Kit (Peqlab) according to the manufacturer's instructions. cDNA was synthesized from 1 μg of total RNA using M-MLV reverse transcriptase (Invitrogen) and qRT-PCR was performed with SYBR Green dye (Biorad). The used qRT-PCR conditions are summarized in Table 2. To determine fold changes in each gene, the housekeeping gene expression of β-actin was used to normalize target gene expression. A list of primer sequences used for qRT-PCR is shown in Table 3.
PPARγ, peroxisome proliferator-activated receptor-gamma; qRT-PCR, quantitative real-time polymerase chain reaction.
Statistical analysis
All experiments were performed with cells isolated from three different animals as biological replicates using at least three technical replicates for each experiment. Data are reported as mean ± standard deviation and analyzed using one-way analysis of variance (ANOVA) or two-way ANOVA followed by Tukey's post hoc tests (GraphPad Prism; GraphPad Software, Inc.). A p-value below 0.05 was considered as statistically significant.
Results
Efficiency of cryoprotection
Preliminary experiments (carried out with small sample volumes in PCR tubes) with and without FBS using varying incubation time (10, 30, and 60 minutes) with CPAs and different DMSO concentrations showed that the highest cell survival could be achieved without FBS included in the tested freezing media. An incubation period of 10 minutes for CPA equilibration resulted in the highest post-thaw survival and was therefore used for the following investigations. In addition, the DMSO content could be reduced to 2.5% when cells were cryopreserved with either MP (see Supplementary Figure S1; Supplementary data are available online at www.liebertpub.com/bio) or MPT (see Supplementary Figure S2), independent of the freezing protocol used compared to control (5% DMSO + FBS).
To verify these findings, the predetermined conditions were used for cryopreservation in cryovials. When cells were frozen with the one-step freezing protocol, no significant differences were found after 24 and 48 hours of recultivation (Fig. 1A) compared to 5% DMSO + FBS. However, a different picture emerged when the cells were frozen with the two-step cooling protocol (Fig. 1B). Under these conditions, a generally higher cell survival was found in comparison to the one-step cooling rate of 1°C/min. After 24 hours post-thaw, a significant difference in recultivation efficiency was seen when cells were frozen with MP − FBS (65%) compared to 5% DMSO + FBS (90%). More cells attached to the culture surface with MPT − FBS (75%). In contrast to the 24-hour result, a significant increase in cell numbers was found after 48 hours of recultivation. After this incubation time, no difference in efficiency was found between the groups (p > 0.05). Therefore, the two-step freezing protocol was used for subsequent studies.
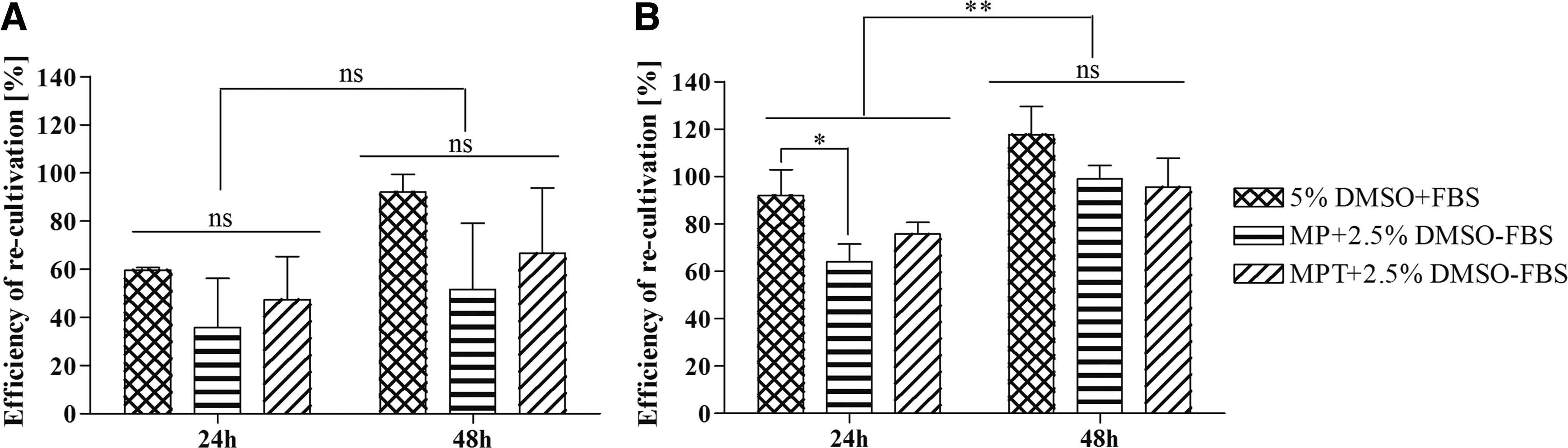
Effect of cryopreservation on the ER of cjMSCs after 24 and 48 hours using different CPAs and a cooling rate of 1°C/min
Metabolic activity
In addition to the ER analysis, the metabolic activity of cells at 24, 48, and 96 hours post-thaw was analyzed (Fig. 2). Up to 48 hours, the activity of frozen cells was significantly impaired (p < 0.05) compared to the nonfrozen control and to 5% DMSO + FBS (p < 0.001, Fig. 2A, B). This trend changed after 72 hours when the MPT − FBS group no longer showed a significant difference compared to nonfrozen cells (Fig. 2C). All cryopreserved groups, including the MP − FBS and MPT − FBS, recovered their metabolic activity after 96 hours (Fig. 2D).
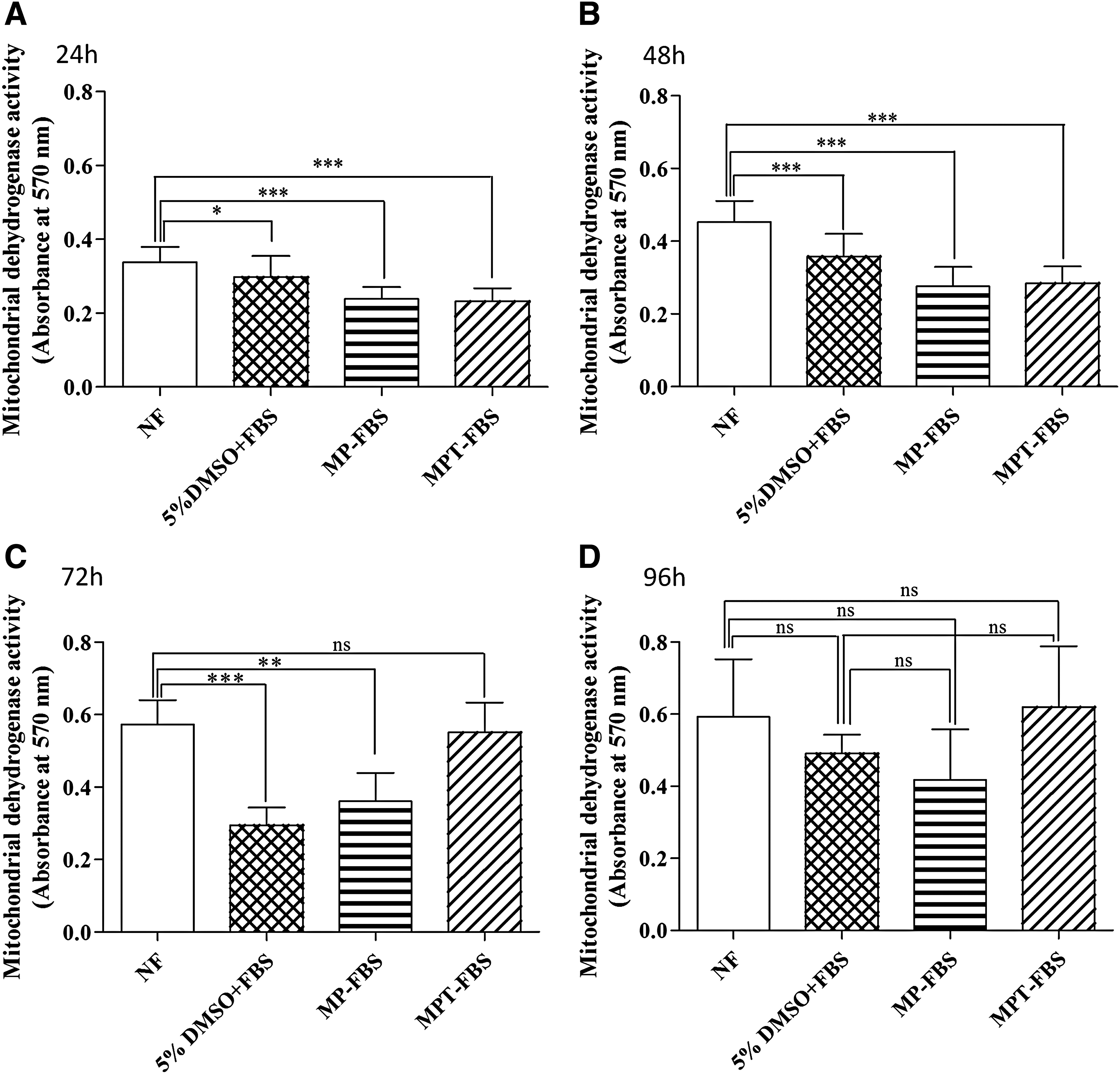
Effect of cryopreservation on the metabolic activity of cjMSCs using different CPAs after 24 hours
Doubling time
After cryopreservation, the doubling time was not significantly different to that of nonfrozen cells. Nonfrozen cells needed ∼28 ± 0.9 hours for one cell division, 26 ± 3.6 hours when frozen with 5% DMSO + FBS, 25 ± 3 hours with MP – FBS, and 26 ± 0.8 hours with MPT − FBS.
Differentiation
Adipogenic differentiation potential
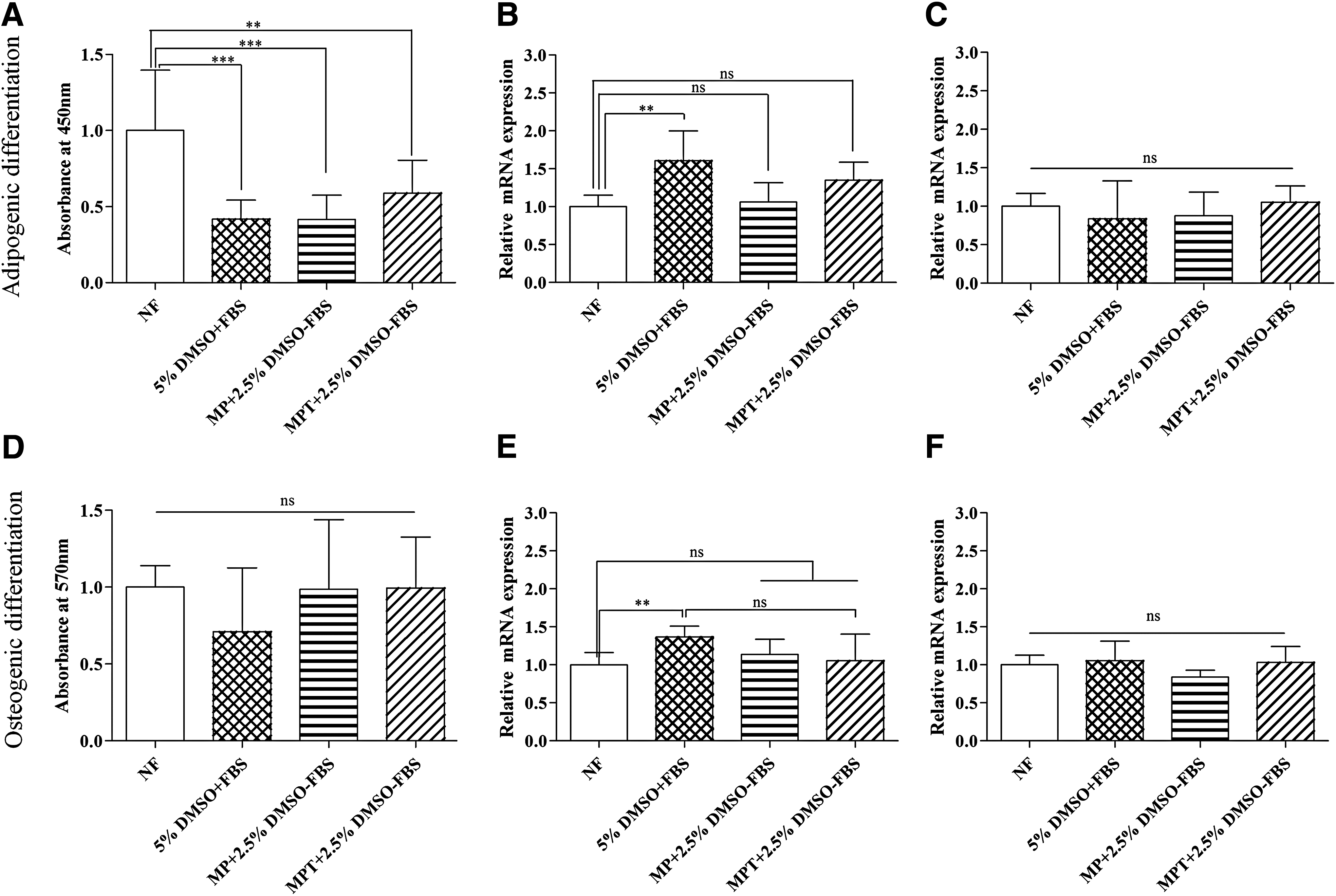
The effect of cryopreservation using different CPAs on the differentiation capacity to adipogenic lineage (Fig. 3) was investigated. Adipogenesis was induced in frozen–thawed BmMSCs, and differentiated cells were analyzed after a 20-day cell cultivation period. BmMSCs were able to form adipocytes with typical fat vacuole formation irrespective of the freezing condition. Oil Red O staining was performed to confirm the presence of fat vacuoles (Fig. 3). Quantification of Oil Red O uptake, however, revealed that there was a significant decrease in oil droplet formation in all of the frozen–thawed differentiated samples compared to fresh unfrozen cells (Fig. 4A). No differences were found between the groups cryopreserved with the three investigated CPA mixtures. Peroxisome proliferator-activated receptor-gamma (PPARγ) and adipsin were chosen to study adipogenic differentiation. The peroxisome proliferator-activated receptors (PPARs) are members of the nuclear hormone receptor superfamily. PPARγ is regarded as the “master regulator” of terminal adipocyte differentiation in mammals.31,32 Adipsin is a mature adipocyte marker. 33 qRT-PCR analysis of adipogenic markers PPARγ and adipsin revealed that there was no significant difference in PPARγ expression across the frozen–thawed samples (p > 0.05) (Fig. 4C). Expression of adipsin was significantly higher in 5% (v/v) frozen cells compared to nonfrozen cells (p < 0.01), but no such significance was found among the MP and MPT groups compared to controls (4B).
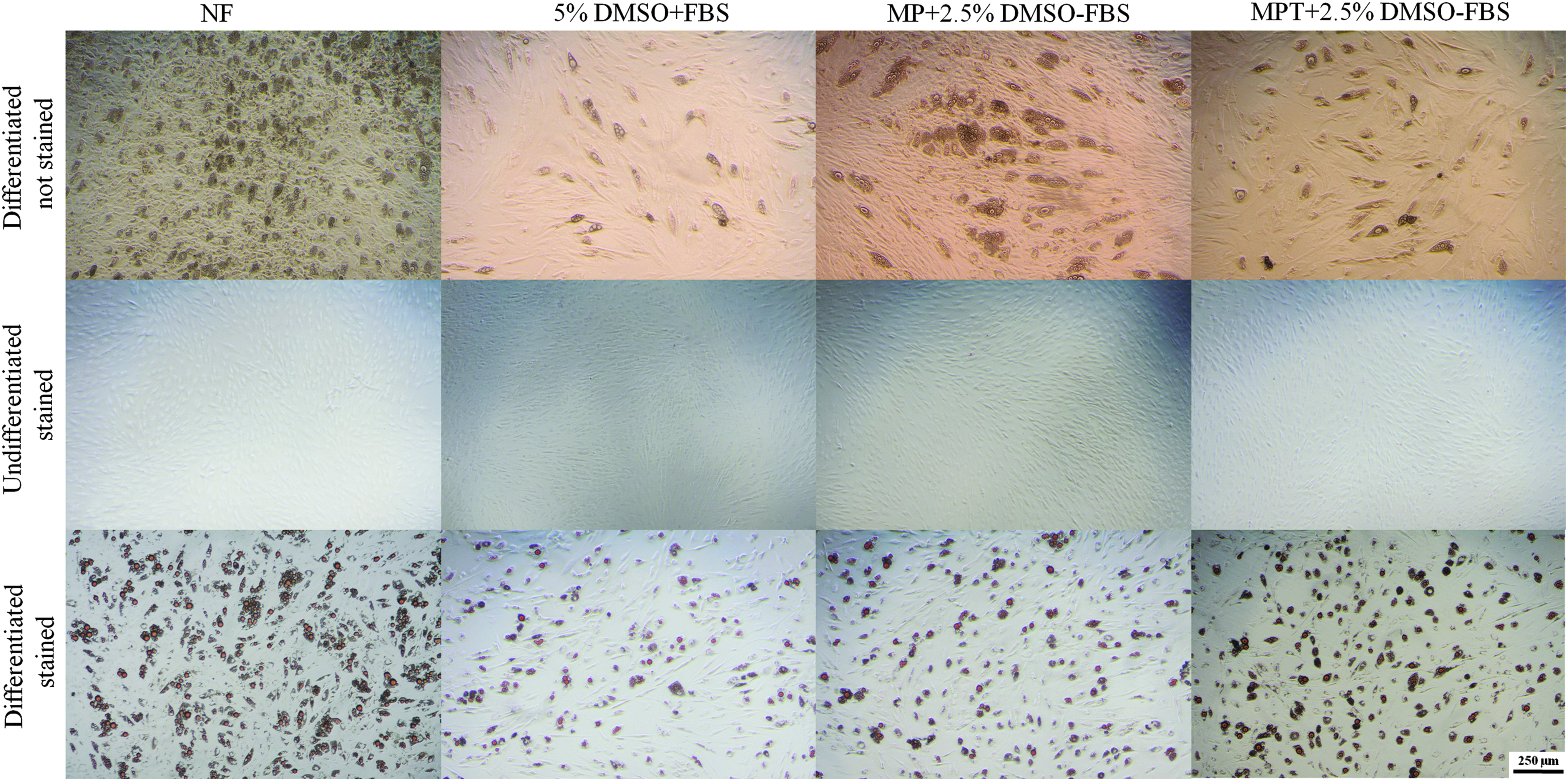
Adipogenic differentiation of cjMSCs after cryopreservation. Representative phase-contrast photomicrographs of cjMSCs cultured under untreated (second row; Oil Red O staining) and adipogenic (third row; Oil Red O staining) conditions. MSCs were differentiated into adipogenic lineage with characteristic fat vacuole formation. The topmost row shows differentiated unstained MSCs on day 20. The middle row shows undifferentiated MSCs stained with Oil Red O. The lowest row shows differentiated MSCs stained with Oil Red O on day 20. Scale bar is 250 μm. Color images available online at www.liebertpub.com/bio
Quantification of Oil Red O uptake
Osteogenic differentiation potential
The effect of cryopreservation using different CPAs on the differentiation capacity to osteogenic lineage was also investigated. Osteogenesis was induced in frozen–thawed samples and differentiated cells were analyzed after a 21-day cell cultivation period. BmMSCs were able to form osteoblasts irrespective of freezing condition. To confirm the osteogenic differentiation of cryopreserved BmMSCs, calcification of extracellular matrix (ECM) was assessed using Alizarin Red S staining on day 21. The presence of reddish staining, indicating abundant calcified ECM, was observed in cells treated with osteogenic media, while the noninduced cells did not stain positively with Alizarin Red S (Fig. 5). These patterns of ECM mineralization were detected in both noncryopreserved and cryopreserved BmMSCs. ECM mineral deposition was reflected by the presence of black nodules using von Kossa staining in both noncryopreserved and cryopreserved samples. During von Kossa staining, darker cellular nodules had more calcium deposition in the ECM. Quantification of Alizarin Red S uptake revealed no significant difference between frozen and nonfrozen samples (p > 0.05) (Fig. 4D). Expression of osteocalcin and osteopontin was assessed with qRT-PCR analysis. Osteocalcin (OCN) is a mineralization-specific extracellular protein forming part of the organic matrix of bone. It is a late bone turnover marker only secreted by terminally differentiated osteoblasts. Osteopontin (OPN) is indicative of osteogenesis. 34 qRT-PCR analysis was performed on day 21 differentiated cultures to evaluate the osteoblastic differentiation parameters of the cryopreserved BmMSCs. The osteogenic induction conditions resulted in the expression of osteocalcin and osteopontin in all the frozen–thawed samples with no significant difference in expression among the cryopreserved samples (p > 0.05) (Fig. 4E, F).

Osteogenic differentiation of cjMSCs after cryopreservation. Representative phase-contrast photomicrographs of cjMSCs cultured under untreated (second and fourth rows; von Kossa and Alizarin Red S staining, respectively) and osteogenic (third and fifth rows; von Kossa and Alizarin Red S staining, respectively) conditions. MSCs were differentiated into osteogenic lineage until day 21. First row (topmost) shows differentiated unstained MSCs on day 21. Second row (from top) shows undifferentiated MSCs stained with von Kossa. Third row (from top) shows differentiated MSCs stained with Von Kossa on day 21. Fourth row (from top) shows undifferentiated MSCs stained with Alizarin Red S. Fifth row (lowest) shows differentiated MSCs stained with Alizarin Red S. No noticeable differences can be observed between all groups and staining methods. Scale bar is 250 μm. Color images available online at www.liebertpub.com/bio
Discussion
In recent years, the importance of biobanks as repositories for biological material has risen significantly. 1 To preserve their viability and functionality, it is imperative to freeze biological samples in a controlled and cell-dependent manner. 1 During cryopreservation, the use of animal serum should be avoided because of immunological reactions and risk of disease transmission to the recipient. As a possible replacement for FBS, MC can be used as a supplement for cryopreservation.6,11 In addition, the “gold standard” CPA DMSO should be replaced or at least reduced to minimal concentrations before clinical use due to its toxic effects. 9 DMSO is known to be toxic to cells and tissues and its toxicity is time, temperature, and concentration dependent. 35 When added to cell culture media, DMSO also induces differentiation of stem cells to cardiac or neuronal-like cells. 36 However, it has been shown to be the most effective CPA for various cell types.35,37
Alternatives such as polymers and sugars are still under investigation. Poloxamer 188 increases viability and has cytoprotective functions, protecting cells against shear stress and mechanical agitation. 21 α-Tocopherol is an antioxidant.16,18 Our data suggested that the cell membrane penetrating DMSO in a concentration of at least 1.5% (v/v) is required. Furthermore, the CPA incubation time played a significant role during freezing (Supplementary Figs. S1 and S2). Higher survival rates were found in the absence of FBS when the CPA combinations of MP and MPT supplemented with 2.5% DMSO were used and when cells were incubated for 10 minutes (Supplementary Fig. S1 and Fig. S2) before freezing. In previous studies it has been shown that antioxidants and apoptosis inhibitors can enhance cell survival, which is also shown in our study (Fig. 1, Supplementary Fig. S1 and Fig. S2).17,18,38,39
After 48 hours of recultivation post-thaw, there was no significant difference between the frozen–thawed samples (Fig. 1). Also, after 96 hours, cells frozen using different CPAs had a similar metabolic activity (Fig. 2). The doubling time of differently frozen samples also did not differ significantly.
Cell functionality of BmMSCs can be proven through trilineage differentiation capacity. The differentiated cells formed fat vacuoles, which could be stained with Oil Red O. The expression of adipsin was lower for MPT (methylcellulose, poloxamer 188, and α-tocopherol, no serum) frozen samples than cells frozen with 5% DMSO (v/v) (Fig. 4B). The frozen–thawed samples could also differentiate to osteogenic lineage with calcium phosphate deposits (Fig. 5). Von Kossa staining revealed brown-black metallic deposits, typical of calcification. Quantitative determination of Alizarin Red S staining showed Ca2+ accumulation in osteogenic cultures. No significant differences could be found between cryopreserved and noncryopreserved BmMSCs cultured under the same condition (p > 0.05) (Fig. 4). There was also no significant difference in expression of osteoblast-specific markers osteocalcin and osteopontin among the cryopreserved samples (p > 0.05) (Fig. 4). Similar results were also shown in previous reports.3,6
Taken together, these results conclusively show that the two alternative freezing media containing a much lower DMSO concentration of 2.5% v/v and no serum can serve as promising freezing media for applications in cryopreservation. Freezing media containing combinations of methylcellulose and DMSO have been reported earlier.3,6 Thirumala et al. used a combination of 10% DMSO (v/v) with 1% methylcellulose and obtained a cell viability of ∼80% for human adipose-derived adult stem cells. 6 The present study, however, suggests that the viability as well as adipogenic and osteogenic differentiation capacity of the post-thawed cells can be maintained using a freezing medium without serum consisting of methylcellulose, poloxamer 188, and α-tocopherol with only 2.5% DMSO. In addition, the chemicals MC and poloxamer 188 have been tested separately for their cryoprotective potential.5,8–10 However, the combination used in the present study has not been reported until now and complements their properties against known cell damage mechanisms during freezing.
Footnotes
Acknowledgments
The authors acknowledge Julia Struss and Jan-Cedric Volbers for their outstanding work and Thomas Mueller for kindly donating multipotent stromal cells. This work has been partially supported by funding from the Deutsche Forschungsgemeinschaft for the Cluster of Excellence REBIRTH (EXC 62/1) and ZIM (Zentrales Innovationsprogramm Mittelstand, KF2654703SB3) in cooperation with Askion.
L.L. and D.S. were involved in the study concept and design, data collection, analysis, and interpretation. L.L. wrote the manuscript with the help of D.S. and N.H. N.H. and B.G. were involved with the study concept and design. A.C. established and provided the staining protocol and primers for osteogenic differentiation and edited the manuscript. B.G. approved the final version.
Author Disclosure Statement
No conflicting financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.