
Abstract

Introduction
B
Materials and Methods
Sample collection
Specimens were collected on September 9, 2014 by NOAA Fisheries Observer Philip Fremont-Smith aboard a commercial fishing vessel on Stellwagen Bank, at the mouth of Massachusetts Bay. The samples were transported on ice for a period of 6–12 hours to the Northeastern University Marine Science Center (Nahant, MA) and were frozen at −80°C upon arrival. Frozen tissue samples (one sample per species, ∼0.5 g ea.) were removed by dissection from the gill filaments of Hippoglossina oblonga and from the musculature of the pelvic fins of Amblyraja radiata and Malacoraja senta and deposited to the Ocean Genome Legacy Genome Resource Repository (accession numbers S22354, S22355, and S22356, respectively). Samples were maintained frozen until use.
DNA extraction
A single subsample (25 mg) was taken from each species' frozen sample for DNA extraction using the Qiagen (Hilden, Germany) DNeasy® Blood and Tissue Kit according to the manufacturer's protocol for purification of total DNA from animal tissues, excluding the optional RNase digest step. DNA extracts were diluted to 10 ng/μL as assessed by the NanoDrop ND8000 spectrophotometer (Thermo Scientific).
Whole genome amplification
One microliter from each species' diluted genomic DNA sample served as the template for WGA using the Illustra™ GenomiPhi™ V2 DNA Amplification Kit by GE Healthcare Life Sciences (Buckinghamshire, United Kingdom) according to the manufacturer's protocol. One microliter of the eluate resulting from a tissueless DNA extraction, performed identically to the experimental treatments except without added tissue, served as a negative control for WGA. As a second negative control, WGA was performed without any added template. Three WGA replicates were performed for each genomic DNA extracted from each of the three species.
DNA barcoding
For each species' genomic DNA sample and three WGA replicates, the “barcode region” of the mitochondrial cytochrome oxidase 1 gene (COI-5P region) was amplified and bidirectionally sequenced using standard protocols. 6 Briefly, 2 μL template DNA, either genomic DNA or WGA product, was used in each PCR reaction. All PCRs had a total volume of 12.5 μL and included: 6.25 μL of 10% trehalose, 2.00 μL of ultra pure water, 1.25 μL 10 × PCR buffer [10 mm KCl, 10 mm (NH4)2SO4, 20 mm Tris-HCl (pH 8.8), 2 mm MgSO4, and 0.1% Triton X-100], 0.625 μL MgCl2 (50 mm), 0.125 μL of each primer cocktail (0.01 mm), 0.0625 μL of each dNTP (10 mm), 0.0625 μL of Taq DNA Polymerase (New England Biolabs), and 2.0 μL of DNA template. The thermocycle profile was: 94°C for 2 minutes, followed by 40 cycles of 94°C for 30 seconds, 52°C for 40 seconds, and 72°C for 1 minute, with a final extension at 72°C for 10 minutes. PCR products were visualized on a 2% agarose E-Gel® 96-well system (Invitrogen) to estimate molecular weight and product concentration. PCR products were labeled using the BigDye® Terminator v.3.1 Cycle Sequencing Kit (Applied Biosystems, Inc.). Each sequencing reaction mixture consisted of 10.0 μL of ultra pure water, 1.0 μL of 5× Sequencing Buffer (Applied Biosystems, Inc.), 1.0 μL of primer (10 μM of M13F or M13R), 1.0 μL of BigDye (Applied Biosystems, Inc.), and 1.5 μL of PCR product. Bidirectional sequencing reactions were performed on an ABI 3730 capillary sequencer.
Bidirectional sequences were assembled from raw trace files using CodonCode Aligner (CodonCode Corporation). Phred scores and length of read (LOR) scores were generated. Primers were manually deleted from sequence contigs. All sequence files were trimmed using the Clip Ends command in CodonCode, using the default parameters. Sample contigs were then manually trimmed to remove sequence data until the bidirectional sequences agreed. To compare WGA sequences to sequences from genomic DNA for each sample, contigs were made from trace files from each species, including WGA replicates. These species contigs were assembled using the Compare Contigs command using the MUSCLE alignment algorithm. 7 DNA sequences were placed in the open reading frame with MEGA version 6.0. 8 These sequences and trace files were deposited in the Barcode of Life Database (BOLD) 9 in the project Whole Genome Amplification (project code WGAP).
Results and Discussion
WGA, PCR, and Sanger sequencing were successful for all samples and replicates. Using a Phred score cutoff of Q > 20, all resulting read lengths were above 550, indicating that 550 or more bases in each sample contig were 99% accurate or greater (Table 1). The average quality scores for all genomic reads and all WGA replicates were 616.33 ± 10.5 and 596.33 ± 7.1, respectively. No significant difference in LOR or average quality score was observed between amplicons from genomic templates and WGA products (Fig. 1). For each of the three species examined, all WGA replicate contigs were identical to their respective genomic templates after editing. Even before manual trimming of consensus reads, the BOLD Identification engine (www.boldsystems.org) returned correct and congruent identifications for all sequenced PCR products, with 100% match to the respective source species observed regardless of the template type used (i.e., genomic DNA or WGA product).
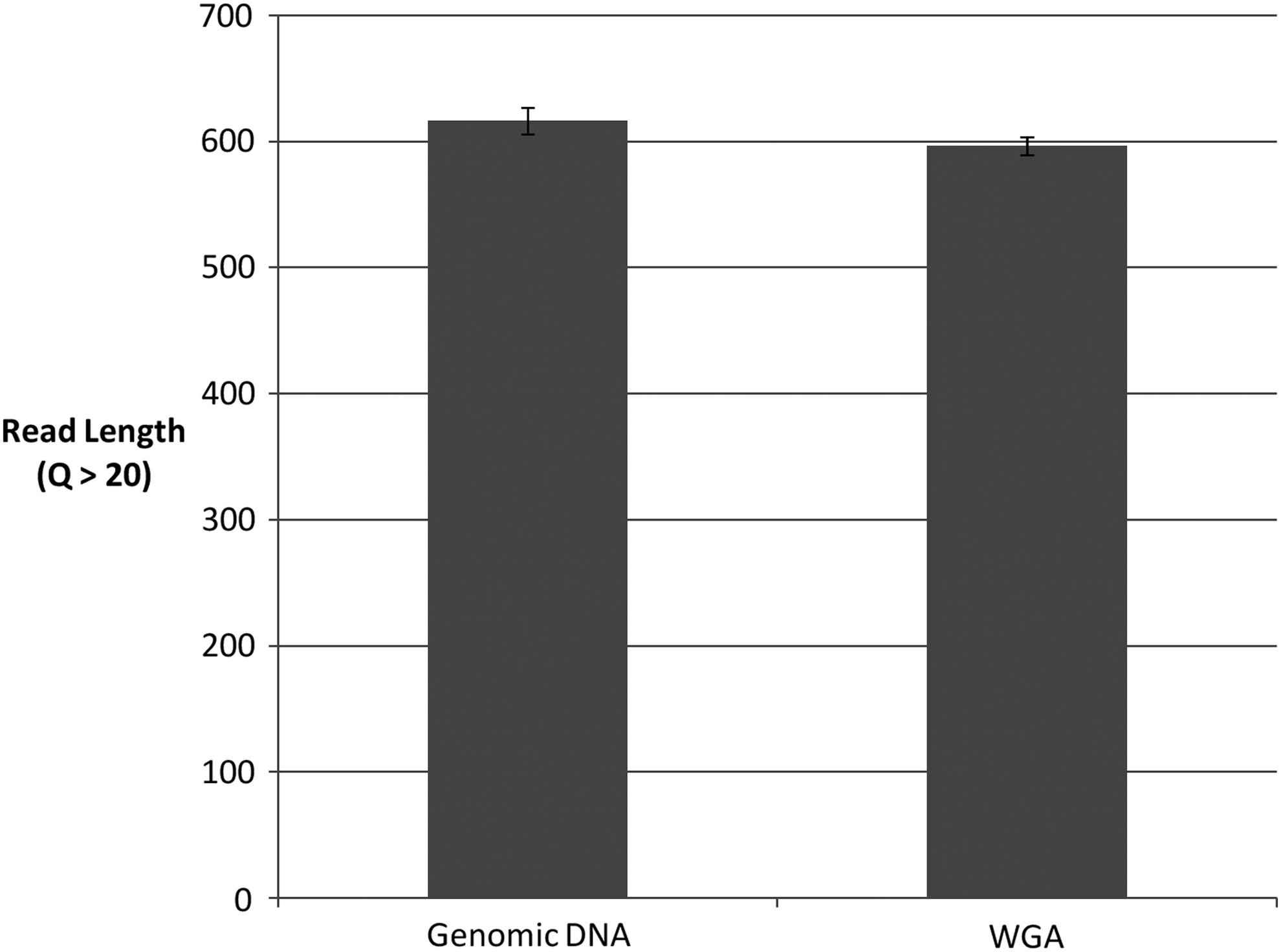
Average read lengths (Q > 20) of genomic and WGA DNA sequences are not significantly different (two-tailed t-test assuming equal variance, p = 0.18). The average genomic DNA read length was 616.33 ± 10.5, and the average WGA read length was 596.33 ± 7.1. WGA, whole genome amplification.
A Phred quality score of 20 indicates 99% base call accuracy for an individual base. The overall Phred20 score indicates the number of bases with Phred scores above 20. In this case, this is also the length of the full edited sequence, indicating that all base calls were over 99% accurate.
WGA, whole genome amplification.
These results indicate that WGA material is as good as genomic DNA for species identification using the DNA barcode region. Although we did not identify any discrepancies, we recommend the use of bidirectional sequencing when conducting identifications to reduce the impact of any potential sequence error. Although these errors would not affect identifications made based on sequence similarity in the case of species identification using DNA barcoding, individual base pair differences can affect other types of identification. The terminal ends contain the greatest probability of sequencing error. 10 We therefore recommend that the terminal 25–50 base pairs of a sequence be avoided when choosing target areas for identification that may be affected by single base differences. 11 This includes design of real-time PCR assays based on DNA barcode sequences and should be considered when developing standards for inclusion in these types of assays.
Presuming a 200 μL eluate from a typical DNA extraction, and 2 μL of DNA required for PCR amplification, performing PCR from reference material would usually allow 100 PCR per extraction. With the WGA protocol used in this study, this can be multiplied 20-fold to at least 2000 reactions; each WGA requires just 1 μL of template DNA and produces 20 μL of product. In theory, WGA product could also be diluted to create even more product; this is a subject for future research. This significantly extends the use of reference material for downstream genetic applications. An alternative method could be to establish cell lines from specimens of interest, but this is a time-consuming, expensive, and impractical venture for biodiversity at large. It is likely that WGA protocols can be optimized to increase the fold of amplification above that used in this study. Furthermore, sequential WGA reactions could effectively provide a limitless source of reference material. Although outside the scope of this short note, further study regarding the possible extension of WGA in this way is warranted.
Amplification and sequencing of the DNA barcode region from genomic template and WGA replicates examined in this study successfully identified the target species. The WGA process did not appear to create erroneous sequence data as an artifact of the process itself. In this analysis, all base call discrepancies arose within terminal ranges by processes also commonly observed in PCR sequencing from genomic DNA templates. 12 We recommend that the terminal bases be avoided in assay design and that these regions be excluded when performing DNA sequence based identification. While our study used the DNA barcode COI region as an example, WGA product could be used to generate sequences from other regions as well, such as nuclear or chloroplast markers. We conclude that WGA is a reliable method of generating biological reference material from limited biospecimen voucher material and that this could be extended for a range of applications.
Footnotes
Acknowledgments
The authors acknowledge the Ocean Genome Legacy Fund at Northeastern University for providing financial support. The authors acknowledge Philip Fremont-Smith for their efforts toward sample collection and the NOAA National Marine Fisheries National Observer Program for providing access to the samples. The authors also thank Timery DeBoer for their help in organizing this project.
Author Disclosure Statement
No conflicting financial interests exist.