
Abstract
Human induced pluripotent stem cell (hiPSC) biobanks are invaluable resources for basic and clinical research, since they provide a sustainable supply of accessible cell lines that meet high quality and safety standards. hiPSCs are particularly useful for understanding disease mechanisms, creating cell models for drug development, and generating novel clinical therapies. For clinical applications and drug discovery, it is fundamental that the acquired pluripotent cell lines never touch animal-derived products nor xenogeneic reagents (Good Manufacturing Practice—grade); whereas for research grade, it is sufficient to operate under Good Laboratory Practice conditions. However, regardless of the end use, it is important that every step in the whole process, starting from the original cells throughout expansion and manipulation, must be performed and recorded rigorously. Here, we describe our biobanking management system that is applied specifically to human pluripotent stem cells.
Introduction
H
A fundamental feature of pluripotent stem cell (PSC) banking is the implementation of a management system that allows the availability of hiPSC collections to the research community as soon as possible after derivation and first publication. This can be achieved through national and international repositories that accept, expand, qualify, and distribute the stem cell lines on a large scale. Distribution requires a series of documents with information regarding the stem cell provenance (acquired through the informed consent) and raw primary data (genomic and functional characterization); quality controls (QCs) and consistency of protocols used in manufacturing. All information should be stored and easily traced. Here, we describe the three levels of QC standardized at ISENET (www.isenet.it). Figure 1 represents an overall map of the different approaches that are undertaken to characterize and validate each hiPSC line and derived bioproduct.

Characterization of produced cell lines and final cell products. Comprehensive map of the different approaches that are implemented at Integrated Systems Engineering (ISENET) to characterize and validate each hiPSC line and its derived bioproducts. hiPSC, human induced pluripotent stem cell.
Level 1 QC: Cell Line Identity and Sterility
The first level of the QC assessment is the basis of the workflow pipeline for the validation of the final cell product. This step includes verification of the hiPSC line's identity and purity, constituting data that add confidence to the results of a scientific study.
Unambiguous and encrypted identification/authentication of cell populations
Cell line misidentification can undermine research results and waste research funds. According to a 2015 economic analysis, it was estimated that $28 billion of irreproducible preclinical research was conducted each year in the United States alone. This financial burden cannot be totally attributed to cell line misidentification but to other factors such as study design and analyses. 11 Very often, researchers are not aware of the existence of a documented standard that codifies specific guidance and laboratory protocols or the availability of authentication services that can make the process relatively easy. Different genomic technologies are available to assess the species origin of each hiPSC line, but only one method is favored and accepted worldwide: short tandem repeat (STR) profiling. STR profiling is an easy and sensitive technology based on polymerase chain reaction (PCR)–based genotyping. It is a reliable method that is routinely used in genetic linkage studies and for paternity identification. 12 More global typing methods, such as single-nucleotide polymorphism (SNP) genotyping, provide a detailed fingerprint of the cell lines, but they are too detailed and expensive to be used for cell identification or authentication. However, additional quality assurance in Good Cell Culture Practice and cell characterization can be implemented. 13
ISENET is a commercial biotechnology firm that, by participating in a European consortium dealing with hiPSC biobanking, developed a workflow program to characterize PSC lines generated by the consortium members. As for the authenticity of the produced lines, DNA genotyping using the AuthentiFiler™ PCR Amplification Kit from Applied Biosystems was adopted. It is a multiplex PCR assay that amplifies nine unique STR loci and the Amelogenin gender-determining marker in a single PCR amplification. On receipt and before creating the master cell bank or seeding stock, STR genotyping is assessed and repeated before storage in liquid nitrogen and distribution as established by the International Cell Line Authentication Committee guidelines (ICLAC: http://iclac.org).
Microbial contamination
It is well known that high cell density culturing of pluripotent and active proliferating cells can encounter bacterial, fungal, mycoplasma, and virus contamination. We routinely apply the guidelines established by European Pharmacopoeia (2.6.27) and Microbial Quality of Pharmaceutical Preparation 5.1.4 (01/2007:50104). However, no current testing regime can guarantee absolute absence of microbial contamination, thus cells remain potentially infectious even after rigorous testing. Besides viral contamination, a variety of other microorganisms can contaminate cell lines, such as mycoplasma. A recent study demonstrated that mycoplasma contamination can significantly alter cellular metabolite levels, confirming the compelling need for routine mycoplasma checking of cell cultures. 14 Although the addition of antibiotics to cultured cells diminishes bacteria contamination, mycoplasma eradication is practically impossible. We check the cell lines for the presence of mycoplasma at least once a month by following ICLAC guidelines.
Optional QC
More detailed QC assays should be performed for those hiPSC lines destined for clinical applications, such as fine-grained detection of microbial and viral contamination through whole transcriptome analysis (WTA). The WTA procedure explores the entire population of RNA transcripts, coding and noncoding, in a quantitative and unbiased manner. Total RNA is ribosomal RNA (rRNA) depleted for removal of cytoplasmic (5S, 5.8S, 18S, and 28S) and mitochondrial (12S and 16S) rRNA, but sample traces of prokaryotic and viral rRNA remain. Then, RNA is sheared and processed for reverse transcription and double-strand complementary DNA synthesis. The final product consists of double-strand DNA molecules of 200–300 base pairs containing copies of the RNAs present in the original sample surrounded by adapters. These molecules are ready for beads amplification at the emulsion PCR step followed by Oligonucleotide Ligation and Detection Sequencing (SOLiD™). An average of 198 million of 50 + 25 nt paired end sequences were produced for each sample.
Genomnia (www.genomnia.com) has developed a computational pipeline for the quantification of microbial reads present at very low levels. After careful rRNA sequence removal, the remaining reads are compared with SILVA, a comprehensive database of annotated 16S rRNAs, 15 counted, and annotated with the available phylogenetic information. An estimation of the microbial contamination can be evaluated on the basis of the rRNA target sequence alignments from the sample reads by using two different strategies: considering all the high-quality alignments to provide a comprehensive overview; or considering the perfect alignments identifying a unique microbial family.
Figure 2 indicates the presence of those contaminants in a longitudinal manner to follow specific microbial families increasing or decreasing over time, in fibroblasts, hiPSC lines, and their differentiated products. When comparing these data with a dataset from published microbial contamination exploration performed by using next-generation sequencing (NGS) on mycobacteria 16 (Sequence Read Archive ID: SRR073723), excellent sensitivity and specificity has been found (Fig. 3). The main advantage of this technique is the unbiased approach: Contamination from microbial families not included in any available kit can be detected and classified from the sequencing data. Moreover, in this case, it is important to define standards in terms of thresholds of identified reads to identify a given cell line as “contaminated” or “potentially contaminated.” 17
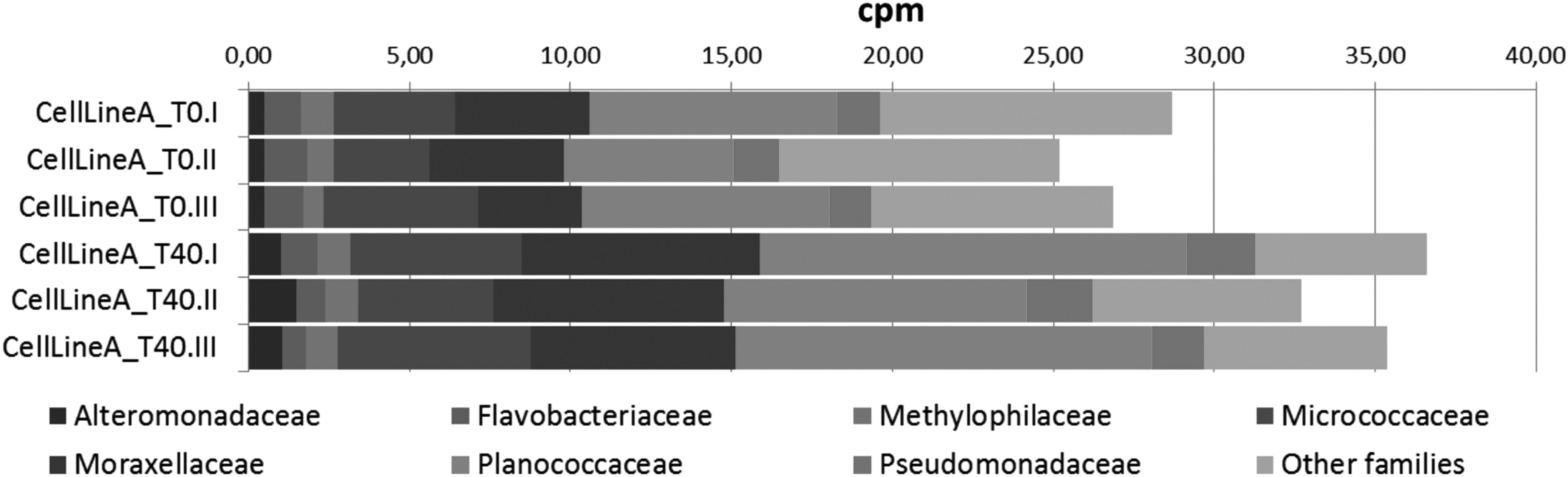
Variations in microbial families' representation in time. Fine-grained detection of microbial contamination performed by WTA. Bar plots report comparisons after induction of differentiation in one representative cell line. I, II, and III represent different sequencing runs. WTA, whole transcriptome analysis.
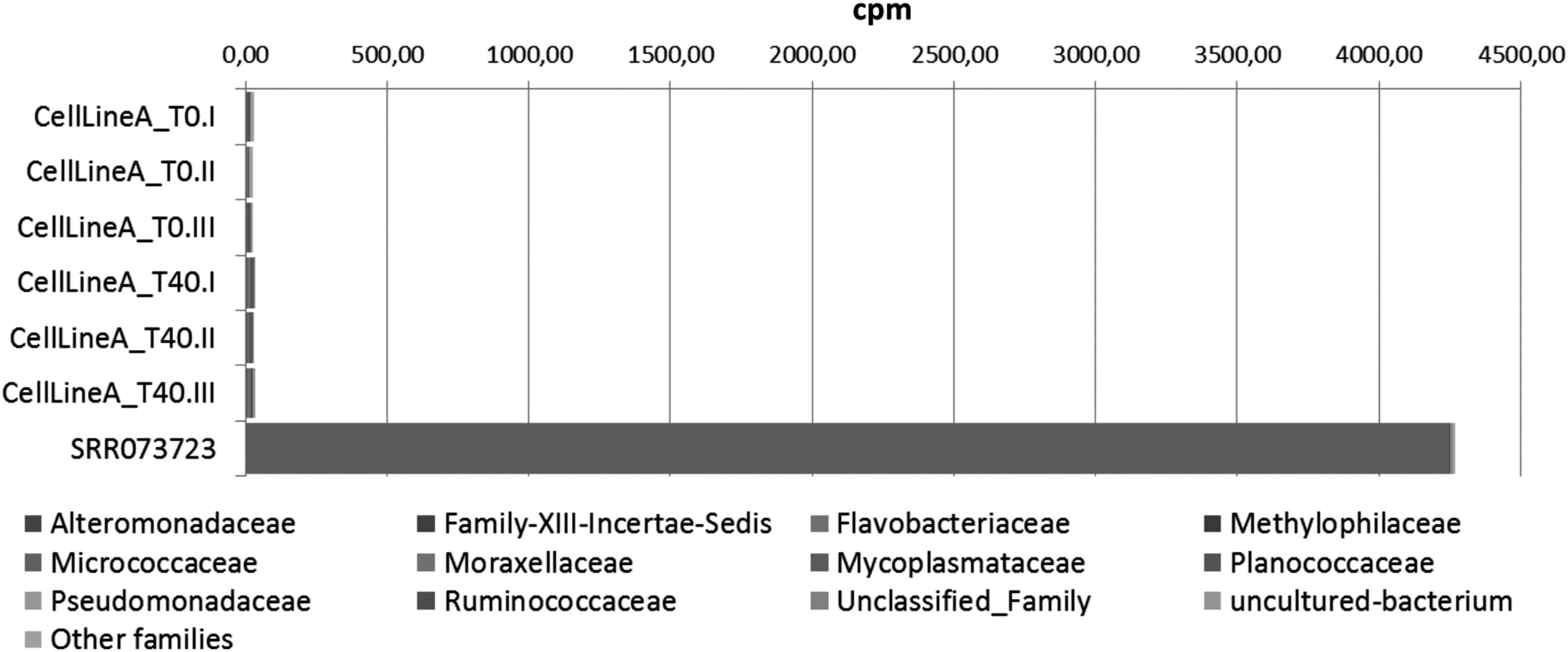
Comparative bacterial analysis by NGS. Results of the contamination analysis pipeline used for different cell types when applied on a mycoplasma-contaminated sample (SRR073723). NGS, next-generation sequencing.
Level 2 QC: Cell Performance and Stability
Genetic stability measurements
On June 9th, 2016/PRNewswire-USNewswire commented on a recent study published in Stem Cell Reports, on a multi-institutional research effort to comprehensively characterize a large set of hiPSCs. The study reported on the genetically unstable and unsafe use of about 30% of the hiPSCs tested due to their instability and contamination from a variable number of undifferentiated cell populations. Technically, each hiPSC colony must be harvested from the plate of origin, free of contaminating original feeder or other undesired cell populations, while retaining the ability to self-renew and, ultimately, to differentiate into any cell type. Table 1 lists measures proposed that are able to detect alterations that may compromise the quality of the lines.
aCGH, array comparative genomic hybridization; FISH, fluorescent in situ hybridization; SKY, spectral karyotyping; SNP, single-nucleotide polymorphism.
Besides using markers associated with undifferentiated cells, such as octamer-binding transcription factor 3/4 (OCT3/4), other factors that can compromise or invalidate the quality of the lines and alter their differentiation potential in an unpredictable manner should be monitored. Telomerase activity, 18 mitochondrial metabolism, genomic stability, and markers of epigenetic change such as DNA methylation (DNAm) and histone modifications are also recommended for those hiPSCs that will be applied clinically. Telomere and telomerase biology play central roles in the indefinite replication and maintenance of the genomic stability of hiPSCs, whereas aberrations in telomere length and maintenance can contribute to cancer development. Research regarding telomere shortening has provided novel insights into the pathogenesis of many diseases, and telomere length measurement has proved to be a powerful tool for the study of many disease processes.
Analysis of chromosome terminal restriction fragments (TRFs) provides the composite lengths of all telomeres in a cell population. In all normal somatic cells examined to date, TRF analysis has shown loss of about 50 to 200 nucleotides of chromosomal telomeric sequence per cell division, which is consistent with the inability of DNA polymerase to replicate the ends of linear DNA. This shortening of telomeres has been proposed to be the mitotic clock by which cells count their divisions, and a sufficiently short telomere may be the signal for replicative senescence in normal cells. In contrast, all immortal cells generally show no loss of telomere length or sequence with cell division, suggesting that maintenance of telomeres is required for cells to escape from replicative senescence and proliferate indefinitely.
Cancer cells have shorter telomeres compared with healthy cells, but they guard their immortality by maintaining these telomeres' length. As conventional in vivo teratoma assays sometimes present drawbacks in their performance, human telomerase reverse transcriptase (hTERT)/telomerase expression levels and telomere length measurement could be considered indicators of tumorigenesis during the hiPSC differentiation. Studies on mouse models suggest that mouse telomerase reverse transcriptase is potentially beneficial as a biomarker, rather than oncogenes of somatic cells, for the assessment of iPSC tumorigenicity. We routinely perform telomere profile analysis as an integrated part of the biorepository workflow to guarantee the availability of high-quality bona fide hiPSC clones and their safe use in research and clinical applications.
Figure 4 shows TERT expression/telomerase activity and telomere length measurement of different hiPSCs compared with human embryonic stem cells (hESCs) H9 and RC17; the latter show long telomeres, high hTERT mRNA expression, and telomerase activity levels that shorten during striatal differentiation (H9_d45). Skin fibroblasts MGM18004E exhibited low and almost-undetectable hTERT expression and activity levels, whereas the fetal neural stem cells (NSCs) CB660SP and neuroepithelial stem cells (NES) AF22 displayed higher transcription levels. The telomere profile of hiPSCs is routinely compared with several cancer cell lines (SH-SY5Y, HepG2, MCF7, GliNS2, and PANC1) to guarantee their genome integrity during cell reprogramming.
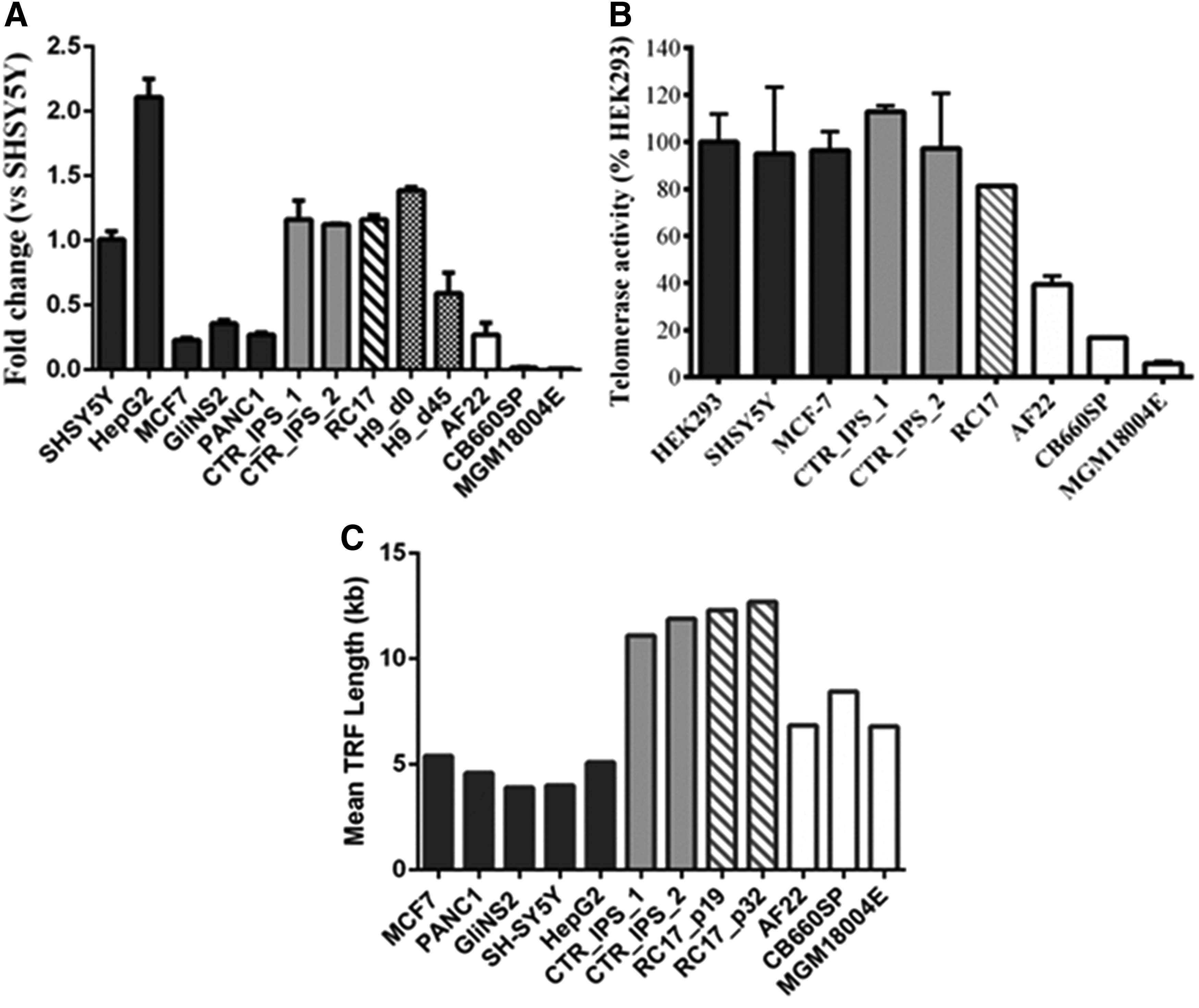
Telomere analysis. The image shows the telomere profile revealed by
Long-term maintenance of stem cells in culture may induce the accumulation of genetic/genomic alterations due to (i) reprogramming methods, (ii) culture conditions, (iii) passaging methods (mechanical vs. enzymatic), and (iv) feeder layer or coating matrix, altogether influencing the stability of each line. Indeed, although new studies on the identification of the best culture conditions have been performed, 19 in vitro expansion still represents a sub-optimal environment compared with the in vivo situation; thus, adaptive genetic changes are expected to occur. The importance of a comprehensive genomic analysis of PSCs is really mandatory, because genetic aberrations may alter disease phenotype, modify functional readouts, as well as impair the well-known delicate balance between genomic instability and carcinogenesis.19–21
Genome integrity can be analyzed with increasing resolution, starting from gross chromosomal abnormalities, such as numerical and structural alterations, to zoom into small copy number variations (CNVs) and point mutations. Conventional karyotyping analysis, including Giemsa (G)- or Quinacrine (Q)-banding techniques, detects alterations >10 Mb in size. Despite the low resolution, it allows the identification of balanced translocations or very low-level mosaicism. Chromosomal microarray analysis, such as array comparative genomic hybridization (aCGH), instead allows the identification of gains and losses of submicroscopic chromosomal material with a resolution of ∼1 Kb in size. The two techniques are not mutually exclusive, rather they are complementary and give distinct information on the genomic composition of the sample. Indeed, despite its higher resolution, aCGH cannot reliably detect mosaicism (not if present in <30% of cells),22,23 and balanced translocations that can be identified instead by karyotype analysis. Results obtained by routine analysis of several hiPSCs clearly suggest that only a combination of different techniques may ensure a reliable coverage of the genomic alterations of the cell lines.
For example, a hiPSC line (PP9#1) derived from a patient with primary progressive multiple sclerosis showed no karyotype abnormalities, whereas aCGH revealed CNVs, including gains and losses spanning from 20 to 700 Kb (Fig. 5A). Furthermore, some identified alterations were polymorphic variants and others, although not polymorphic in the population, were not associated with any clinically significant phenotype. Moreover, karyotyping analysis on a control fibroblast cell line (F0075) shows a 46,XX[40];46,XX,-4,+mar[10] profile, which is an index of a low level of mosaicism (20%), which is probably derived from a rearrangement of chromosome 4 (Fig. 5B). Conversely, aCGH did not reveal any alteration, probably for two reasons: The alteration did not involve gain or loss of genomic material, and/or the level of mosaicism was below the detection sensitivity threshold. 24

Karyotype analysis. The image shows the complementarity of karyotype analysis between Q-Banding by Fluorescence using Quinacrine (QFQ-banding) (upper panel) and aCGH performed by SurePrint G3 Human CGH Microarray Kit 60K (Agilent) (lower panel).
CNVs SNPs microarray analysis is the ideal technology to assess the chromosomal stability of clinical-grade hiPSCs. Deleterious CNVs can arise during the reprogramming process and can result in clonal selection advantages that are undesirable for further applications. Further, such aberrations can result in the deletion of known tumor suppressors or duplication of cell growth/oncogenic factors, necessitating a more comprehensive analysis of the genomic stability of the reprogrammed cell lines.
Level 3 QC: Cell Product Characterization
Measure of heterogeneity and differentiation ability
hiPSCs hold a delicate balance between pluripotency and differentiation, and they cannot be maintained in an entirely homogeneous state, even in the most careful laboratory. Moreover, the same cell line, even when cultured in apparently identical conditions, accumulates different stochastic changes. Furthermore, different cell lines generated from the same donor can differ in phenotype because of allelic differences due to culture adaptation. In addition, different laboratories may have different degrees of tolerance for acceptable differentiation or degree of feeder contamination. It is, therefore, mandatory to define standards of expression markers in the undifferentiated state as well as to evaluate the presence of differentiation markers that are responsible for cell heterogeneity and complex readout.
Table 2 describes useful assays to assess pluripotency features and spontaneous differentiation capacity. The most accessible methods in the laboratories are immunocytochemistry and flow cytometry staining. Antibodies generated for epitopes such as stage-specific embryonic antigen-4 (SSEA-4) and keratan sulfate antigens TRA1–81 and TRA1–60 are widely used to characterize PSCs, whereas novel epitopes are continuously being identified. Flow cytometry measurements for TRA markers should be above 90%, whereas stage-specific embryonic antigen-1 (SSEA-1) expression is below 5%. During reprogramming, partially reprogrammed colonies can arise early but are unable to generate full PSCs and not all clonal lines have an equal capacity to differentiate into desired final cell products, in vitro.
FACS, fluorescence-activated cell sorting; RT-PCR, reverse transcription–polymerase chain reaction; qPCR, quantitative PCR; SELDI-TOF, surface-enhanced laser desorption/ionization time-of-flight.
Further, given that the efficiency of reprogramming remains overall poor (∼0.001% to 1%) (www.stemgent.com/applications/reprogramming), it is reasonable to apply criteria to select clones with a better pluripotency potential based on live fluorescent staining for alkaline phosphatase (AP), or to enrich for cells that express high levels of recognized surface stemness markers such as polysialylated-neural cell adhesion molecule (PSA-NCAM)+ and CD133+.25,26 AP activity is upregulated in hiPSCs and is often used to distinguish them from feeder and parental cells during reprogramming or during early screening, to determine which colonies should be selected for further culture. This will help to synchronize and homogenize the cell culture. Moreover, the gold standard of pluripotency is the teratoma assay combined with histopathological analysis.
Optional QC
There are a number of other test types that can further control and qualify the differentiated cell products, such as whole transcriptome, methyl-seq analysis, miRNA expression, or the combination of all techniques. Collection of these types of data is considered optional but would become crucial if the hiPSCs are used in clinical or pharmacological settings. Individual histone modification and DNAm are the most common measures of epigenetic changes. Both types of analysis are labor intensive, but array-based methods have the potential to allow for inexpensive assessment of the methylation status of hundreds of regulatory elements, on a relatively large scale, with a good tradeoff between quality and computational time when compared with high-throughput large-scale data collection and respective analysis.
On the other hand, high-throughput methods such as NGS offer genome-wide, unbiased, and highly sensitive views. Moreover, hiPSCs hold a strong memory of their somatic origin and reprogramming, independently of the applied technology, and they do not completely erase the original cell fingerprint.27–30 Cell memory can be assessed by performing global methylation analysis both before and after reprogramming, as well as on the final cell product.
We perform DNAm analysis only on a limited number of cell lines and compare the profiles of the undifferentiated cells with their final cell products, focusing on the identification of novel markers of differentiation and for bio-security. DNAm is one of the most studied epigenetic modifications, and its importance increases during embryonic development and cell differentiation. Therefore, the use of DNAm profiles could be considered a valuable tool in the classification of pluripotent and nonpluripotent cells. 31
Moreover, DNAm changes also appear during senescence and cancer; thus, the investigation of these differences may complement the QC of hiPSCs, becoming a sort of “stand alone level of QC.” DNAm changes may contribute to the genomic stability of pluripotent cells and to their ability to differentiate into specific cellular identities. The investigation of DNAm profiles might improve the knowledge of pluripotent cell differentiation capacity and their tumorigenic potential considering their future use in drug screening, disease modeling, and regenerative medicine. 32 All these observations highlight the importance of using different levels of QCs to determine both the experimental reliability and biosafety of pluripotent-derived cellular products. PSCs have peculiar DNAm signatures that mirror their developmental potential. hiPSCs are characterized by DNAm profiles that resemble those of hESCs, although the former retain a sort of epigenetic memory of their tissue of origin.27–29
We investigated DNAm profiles of disease-specific and control hiPSCs in different stages of differentiation to final committed products using an array-based genome-wide platform. 30 This approach enabled the establishment of a reference database, which is valuable for studies dealing with the identification of DNAm changes occurring during cell fate commitment. DNAm changes may also contribute to cancer initiation, through both hyper-methylation of tumor suppressor genes and/or hypomethylation of oncogenes and genomic instability.31,32 The comparison of DNAm changes between differentiating and cancer cells could identify new methylation-based safety biomarkers.
A recent study on hESCs revealed a tumor suppressor gene, BCL2L11, with a hypermethylated promoter, which resulted in its RNA down-modulation during striatal neuron differentiation. 30 Consequently, these findings unveil the hurdles in the evaluation of the relative risk–benefit ratio of cell-based products. Risk assessment and the identification of methylation-based biomarkers are still challenges in the pluripotent cell field. Nonetheless, DNAm changes are subtle and are influenced by several extrinsic factors. In any case, nowadays, the real bottleneck is the small number of datasets available to establish a reliable standard in biomarker discovery and to distinguish between potentially dangerous and nonadverse signals for cellular biosafety.
Figure 1 describes a comprehensive map of the multidisciplinary technical approach to thoroughly qualify the final stem cell product and track the potential source of variations that may occur among the different cell lines (Table 3). The database should contain information related to the generated hiPSC lines, such as donor medical history, ethnic background, age, gender, drug treatment, specimen availability, technological intervention, and several phenotypic outcomes. The database should be managed by the biobank that releases an elaborated “scorecard” for each hiPSC line and all derived cell products. To properly set up comparative and informative experiments among the different hiPSCs, it is important to standardize the expansion procedures and use reagents for identical tests with stocked materials.
Conclusions and Future Perspectives
An increasing number of diseased cell types are being created by human induced pluripotent stem cells (hiPSCs) year after year. However, many limitations are encountered and need to be overcome, among which are qualification and biobanking processes. Based on these limitations, the present situation of the hiPSCs' applications deals with the laboratory-scale production and testing assay. hiPSCs need to be systematically controlled and characterized to increase their therapeutic potential. By using integrative analysis across genomic platforms, we focused our efforts on studying hiPSC phenotype, their genetic/epigenetic status, and gene regulation. We applied a combination of different stringent assays to characterize a number of human pluripotent stem cells (hPSCs) and their derivative final products (Fig. 1), which will serve as a valuable resource, as hiPSC technology moves into clinical translation.
Footnotes
Acknowledgments
The authors acknowledge the grants from NeuroStemcellRepair (European Union Seventh Framework Programme, Grant Agreement No. 602278), MIUR Regione Lombardia Network Lombardo iPS (NetLiPS, Project ID 30190629-2011), Progetto Quadro Regione Lombardia-CNR (RSPPTECH 2013–2015), and InterOmics Flagship Project (2015), which allowed them to develop an integrated platform to characterize human pluripotent stem cell lines.
Author Disclosure Statement
P. DB. is ISENET's CEO, and I.B. is the Scientific Director. A.M. is GENOMNIA's Scientific Director, and A.G. is responsible for the bioinformatics unit.