
Abstract
Cryopreservation includes a set of techniques aimed at storing biological samples and preserving their biochemical and functional features without any significant alterations. This study set out to investigate the effects induced by cryopreservation on cultured sheepskin fibroblasts (CSSF) through cryomicroscopy and gene expression analysis after subsequent in vitro culture. CSSF cells were cryopreserved in a cryomicroscope (CM) or in a straw programmable freezer (SPF) using a similar thermal profile (cooling rate −5°C/min to −120°C, then −150°C/min to −196°C). CSSF volume and intracellular ice formation (IIF) were monitored by a CM, while gene expression levels were investigated by real-time polymerase chain reaction in SPF-cryopreserved cells immediately after thawing (T0) and after 24 or 48 hours (T24, T48) of post-thaw in vitro culture. No significant difference in cell viability was observed at T0 between CM and SPF samples, while both CM and SPF groups showed lower viability (p < 0.05) compared to the untreated control group. Gene expression analysis of cryopreserved CSSF 24 and 48 hours post-thawing showed a significant upregulation of the genes involved in protein folding and antioxidant mechanisms (HPS90b and SOD1), while a transient increase (p < 0.05) in the expression levels of OCT4, BCL2, and GAPDH was detected 24 hours post-thawing. Overall, our data suggest that cryostored CSSF need at least 24 hours to activate specific networks to promote cell readaptation.
Introduction
C
The fate of cells in relation to IIF or dehydration is closely related to the cooling rate. In fact, slow cooling leads to dehydration, while fast cooling causes IIF. 1 The consequences include solution osmotic effects due to phase changes affecting water during cooling and thawing, 3 toxic effects due to cryoprotectant (CPA) concentration, 4 and mechanical effects generated by the growth of ice crystals.1,3 From a biological perspective, this implies a reduction in viability, oxidative stress, biochemical modifications, deferred apoptosis, and cytoskeletal disassembly. 5 The final consequences of these injuries are reduced viability and/or alterations in cell function that are yet to be fully clarified.
The first methods used to evaluate the effects induced on cryopreserved cells by exposure to a very low temperature are mainly based on the detection of cell membrane integrity,2,6–10 measuring metabolic activity,11–13 and proliferation rates. 14 However, these aspects do not provide a complete picture of the complexity of the reactions triggered by cryopreservation; therefore, the evaluation of additional parameters is required.
Several types of cells are subjected to cryopreservation, and wide attention has recently been paid to stem and reproductive cells.9,14–18 In relation to somatic cells, our knowledge on the cryopreservation of primary fibroblast cultures derived from skin biopsy lacks basic information regarding cryo-induced molecular changes that can affect further use. Indeed, these cells find potential application in several fields. They may be employed in the development of induced pluripotent stem (iPS) cells 19 and cloning through somatic cell nuclear transfer (SCNT), 20 which would allow the preservation of the genomes of endangered species.21,22 Because of their involvement in wound healing, these cells may be used in the creation of artificial engineered tissues both for medical skin implants and in drug development, in chemical and cosmetics tests.2,4,12,13 Cultured skin fibroblasts also find clinical application in aesthetic surgery 23 and have recently been employed for somatic cell phenotype switch through the use of epigenetic modifiers.24,25
The possibility to view the events that occur in the cells during the freezing procedure in real time may contribute to our understanding of cell response to exposure to very low temperatures (−196°C), facilitating the definition of an optimal cryopreservation profile. Modern cryomicroscopes (CM) 26 allow the investigation of the biophysical changes that take place during cryopreservation, recording cell volume variations determined by cell dehydration7,8,27 and the dynamics of IIF.28–32 The evaluation of the parameters related to temperature and cooling/warming rates may lead to define a biophysical model, which could be helpful in improving cryoprotocols. 28 Studies on cryopreserved fibroblasts have generally considered biophysical parameters with the aim of modeling the real behavior of cells in defined cryopreservation conditions7,33,34
On the other hand, the relative quantification of gene expression may provide more in-depth information regarding the functional changes induced on the cell by cryopreservation. As it is known, cell freezing triggers the expression of a set of genes involved in the recovery of cell damage, apoptosis, and response to thermal stress. 16 Thus, we selected a panel of genes potentially affected by cryopreservation (HSP90b, CIRBP, and KIF11/Eg5),16,35,36 involved in apoptosis (BAX, BCL2, and GAPDH),14,37–42 and oxidative stress (SOD1). 37 Moreover, we included a marker generally associated with pluripotency (OCT4), whose expression was detected at the basal cell level in somatic cells 11 and was seen to vary in response to heat shock or cryopreservation.43,44 The relative quantification was performed immediately after thawing (T0) and after 24 or 48 hours (T24, T48) of post-thaw in vitro culture, as at the moment only a few studies investigated the levels of messenger RNA (mRNA) in cryopreserved fibroblasts after the thawing process.13,36
Sheep have been used as a model since the first experiments involving fibroblast cells in SCNT were performed. 20 Research on this species found application in several medically relevant fields, 45 but most information involving cryopreservation concerns cardiac valve graft, 46 respiratory medicine, 47 orthopedic, 48 and reproductive issues. 49
This work is aimed at studying the effects induced by cryopreservation on in vitro-cultured sheepskin fibroblasts (CSSF) derived from ear biopsies in terms of morphology and gene expression changes. For this purpose, volume modification and IIF were examined through optical cryomicroscopy by setting a definite cooling thermal profile. Furthermore, the expression levels of a panel of genes were relatively quantified by real-time polymerase chain reaction (PCR) in cryopreserved CSSF, immediately after thawing and after 24 and 48 hours of subsequent in vitro culture. The results might help improve cryopreservation protocols and optimize post-thawing manipulation.
Materials and Methods
Skin fibroblast cells: isolation and culture
All operations were performed in the laminar flow hood. Skin fibroblasts were obtained through an ear biopsy of a slaughtered adult sheep (in laboratory within 3 hours). The ear was washed with 70% ethanol, cut, and put into a sterile tube with a collection medium consisting of alpha-minimum essential medium (α-MEM) and 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES; Life Technologies).
The biopsy was placed into a sterile Petri dish with 1 mL of sheep heat-inactivated serum and cut into small pieces (1 mm3) with scissors and a scalpel. The pieces were seeded in a new 60 mm tissue culture Petri dish and left to adhere in an incubator for 30 minutes. Then 5 mL of α-MEM culture medium supplemented with 10% (v/v) fetal bovine serum (FBS; Life Technologies), 1% glutamine (v/v) (Sigma), and 1% (v/v) penicillin/streptomycin (Sigma; stock 5000 U/mL, 5000 μg/mL) was added. The culture was kept at 37°C, 5% CO2, and 100% humidity. The medium was changed after 5–7 days. Once the culture was almost confluent, it was detached with trypsin 0.25% and ethylenediaminetetraacetic acid (EDTA) 0.04% (v/v; Sigma) and split until the fifth passage (P5), when the experiments were performed.
Immunocytochemistry
To characterize the homogeneity of the isolated cell population, we evaluated the presence of vimentin, the typical fibroblast intermediate filament protein, by immunocytochemistry.24,25 The cultured cells (P5) were washed three times with phosphate-buffered saline (PBS, Triton 0.1%), fixed with chilled absolute methanol, and permeabilized for 1 hour (PBS, Triton 0.1%, bovine serum albumin 3%). Anti-vimentin primary antibodies (M 0725; DakoCytomation) were applied overnight at 4°C (working dilution 1:300 in permeabilization solution). The cells were incubated with suitable secondary antibody (AlexaFluor 488; Life Technologies) for 1 hour. The nuclei were stained for 15 minutes with Hoechst (2 μg/mL; Sigma). All samples were observed under a Leica TCS SP5 confocal microscope.
Cellular viability
Viability was assessed after each detachment from the Petri dishes (uncryopreserved cells) and after thawing by analyzing cell membrane integrity with Trypan blue staining. Briefly, a 1:1 dilution of cell suspension was prepared using a Trypan blue solution (0.4% Trypan blue in PBS, w/v) and 10 μL was loaded in a hemocytometer. Nonviable cells were blue and viable cells were unstained. The survival rate (SR) was calculated as the ratio of the number of unstained cells to total number × 100.
Cell growth curve
Growth curve evaluation was carried out by plating 2 × 103 cells/well in 24-well multidishes (Nunc). The number of cells was counted using a hemocytometer. Cell viability was determined by the Trypan blue dye exclusion assay. Both control and cryopreserved groups were analyzed.
Sample preparation
When the cell culture reached 90% confluence (P5), the culture plates were washed once with PBS Ca++/Mg++ free, trypsinized for 5 minutes, centrifuged for 4 minutes at 170 × g, and resuspended in the complete medium. After the count, the cells were centrifuged for 2 minutes at 170 × g and resuspended in cryopreservation solution (50% α-MEM, 40% FBS, 10% dimethyl sulfoxide [Me2SO]); equilibration was performed for 15 minutes at room temperature.
Cryopreservation
A common thermal profile was set up with two systems of controlled-rate freezing: a CM mounted on a BCS196 stage (Linkam, Tadworth, United Kingdom) and a CL-8000 straw programmable freezer (SPF; Cryo Logic, Mount Waverley, AU). All experiments were performed in the absence of ice seeding and nucleation occurred spontaneously.
IIF and variation in cellular volume were investigated with the CM. The cells to be used for gene expression analysis were cryopreserved with the SPF. After cryopreservation with both methods, cell viability was evaluated immediately after thawing (T0).
Cryomicroscopy studies
The CM consists of the following: BCS196 cryostage, imaging station with all items supplied, PE95/T95 system controller, and Linksys32 software. Temperature-controlled range varies between +125°C and −196°C, with a maximum heating rate of 150°C/min and a minimum of 0.01°C/min.
A small drop of cell suspension (0.005 mL × [2.4 × 106/mL]) was loaded onto the center of a Quartz Crucible and this was gently closed on the top with a 13 mm cover slip. The entire system was placed onto the silver block and the cryostage was closed. Before starting, the air was purged by filling the stage chamber with dry nitrogen, to eliminate the moisture that can condense or freeze inside the chamber and hinder image quality. Data capture and real-time monitoring were performed with a Qimaging camera (Qicam fast 1394 CCD) using a 20 × long-distance objective lens.
The temperature profile started at room temperature (20°C) and was planned in four steps: (a) the sample was cooled at −5°C/min to −4°C and kept at this temperature for 3 minutes to adjust the focus and optical setting of the Linksys32 software, (b) then it was cooled to −120°C at −5°C/min, (c) cooled faster at −150°C/min to −196°C, and held for 10 minutes before (d) being warmed to 20°C at +150°C/min. Overall, 14 experiments were performed and 195 cells were observed.
Light images for IIF studies were recorded at 1 frame per second (fps) between −4°C and −35°C, as we expected to observe the EIF in the samples within this temperature range. Video data analysis was performed observing, in every image frame, the number and size of cells in which darkening (a sudden blackening of the cells) occurred. The probability of intracellular ice formation (PIIF) in cells was calculated as the ratio of the cumulative number of cells exhibiting darkening to the total number of cells in the sample population.
Cell volume was evaluated for each experiment through single image frames collected at defined temperature values before the occurrence of ice formation: +15°C, +4°C, 0°C, −4°C, −10°C, and −20°C. Only static cells with a spherical shape were considered. Cellular volume was calculated according to the formula
SPF studies
One freezing protocol was carried out with the SPF made by Cryo Logic and equipped with the CL8000 temperature controller. The working temperature ranged between +40°C and −120°C. The cell suspension was diluted in cryopreservation solution (0.21 mL × [1 × 106/mL]) and loaded into straws. The thermal profile was constructed in four steps: (a) starting at 20°C, cooling at −10°C/min to −4°C, and holding for 3 minutes, (b) cooling down at −5°C/min to −120°C, (c) pulling out the straws from the cryochamber and plunging them into liquid nitrogen for 10 minutes, and (d) thawing in a warm bath (37°C) until all ice crystals were melted. Ice nucleation occurred spontaneously without ice seeding. After thawing, the CPA was removed from each straw by dilution in the culturing medium (1:9 v/v, 37°C for 15 minutes). Each diluted cell suspension was split into two aliquots: the first was immediately centrifuged at 170 × g for 4 minutes and the pellet was stored at −80°C for the following molecular analysis, and the second one was put in a Petri dish, cultured in vitro for 24 or 48 hours at 37°C, 5% CO2, 100% humidity, and then collected for further analysis.
Gene expression analysis
RNA samples were isolated from the skin fibroblasts cryopreserved in the SPF and/or in vitro cultured after cryopreservation.
Experimental groups:
• T0 CTR: control fibroblasts (not cryopreserved) • T0 CRYO: cryopreserved fibroblasts, analyzed immediately after thawing • T24 CTR: control fibroblasts cultured in vitro for 24 hours • T24 CRYO: cryopreserved fibroblasts, analyzed after 24-hour post-thawing in vitro culture • T48 CTR: control fibroblasts cultured in vitro for 48 hours • T48 CRYO: cryopreserved fibroblasts, analyzed after 48-hour post-thawing in vitro culture.
The cells (≈5 × 105) were stored in an RTL buffer (RNeasy Micro Kit; Qiagen, Hilden, Germany) at −80°C until further analysis. Three replicates for each experimental group were analyzed.
RNA isolation and reverse transcription
The total RNA was isolated from cells with the RNeasy Micro Kit (Qiagen) following the manufacturer's instructions. Five picograms of luciferase mRNA (Promega) were added to each group before RNA extraction to account for RNA loss during the isolation process.
During the procedure, the RNA was treated with DNase I to exclude any potential genomic DNA contamination.
The RNA was quantified by NanoDropLite spectrophotometer (Fisher Scientific S.A.S.). Forty nanograms were used for complementary DNA (cDNA) synthesis by reverse transcription–polymerase chain reaction (RT-PCR).
The reaction was performed in a final volume of 20 μL, consisting of 50 mM Tris–HCl (pH 8.3), 75 mM KCl, 3 mM MgCl2, 5 mM DTT, 1 mM dNTPs, 2.5 μM random hexamer primers, 0.05 μg oligo (dT)18 primers, 20 U RNase OUT, and 100 U SuperScript III RT (all purchased at Invitrogen Corporation, Carlsbad, CA). The reaction tubes were incubated at 25°C for 10 minutes, then at 42°C for 1 hour, and finally at 70°C for 15 minutes to inactivate the reaction. One tube without RNA and one with RNA, but without reverse transcriptase, were analyzed as negative controls. To quantify the RNA recovery rate, 5 pg of luciferase mRNA (not subjected to RNA isolation) were subjected to cDNA synthesis as well.
Real-time polymerase chain reaction
Primers for all genes studied are listed in Table 1. Relative quantification of transcripts was performed by real-time polymerase chain reaction (RT-PCR) in a 7900HT Real-Time PCR System (Applied Biosystems, Foster City, CA). PCR was performed in a 15 μL reaction volume containing 7.5 μL 2× SYBR Green PCR Master Mix (Applied Biosystems), 200 nM of each primer, and cDNA equivalent to 5 ng RNA.
Ta, annealing temperature.
The PCR protocol consisted of two incubation steps (50°C for 5 minutes and 95°C for 2 minutes), followed by 40 cycles of amplification program (95°C for 15 seconds, gene-specific annealing temperature [Table 1] for 30 seconds, and 72°C for 30 seconds), a melting curve program (65°C–95°C, starting fluorescence acquisition at 65°C and taking measurements at 10-second intervals until the temperature reached 95°C), and finally a cooling step to 4°C. Fluorescence data were acquired during the 72°C extension steps.
To minimize variations due to handling, all samples to be compared were run on the same plate using a PCR master mix containing all reaction components apart from the sample.
The sizes of the RT-PCR products were further confirmed by gel electrophoresis on a 2% agarose gel stained with Sybr Safe (Invitrogen Corporation) and visualized by exposure to blue light. The PCR products were sequenced (Model 3130xl Genetic Analyzer; Applied Biosystems) after purification with the MinElute PCR purification kit (Qiagen) and sequence identities were confirmed with BLAST (www.ncbi.nlm.nih.gov/BLAST).
The relative quantification of gene expression was performed with the 2-ddCq method. 50 The expression analysis was performed normalizing each target gene against the geometric mean of four internal reference genes (succinate dehydrogenase complex subunit A [SDHA], tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein zeta [YWHAZ], ribosomal protein L9 [RPL9], and actin B [ACTB]). Before normalization, the stable expression of the reference genes among groups was assessed.
Statistical analysis
The data were analyzed with the MINITAB Release 12.1 software package (Minitab, Inc., State College, PA).
The viability data were examined by analysis of variance (ANOVA) followed by Tukey's post-hoc comparisons. After testing for normality and equal variance using the Kolmogorov–Smirnov and Levene tests, respectively, the transcript data were analyzed with General Linear Model ANOVA, followed by Tukey's post-hoc comparisons when p values were significant. Differences were considered significant when p < 0.05.
Results
Cell viability, immunocytochemistry, and cell growth curve
Membrane integrity was tested immediately after thawing (T0) in both samples processed with CM and SPF, in samples recultured for 24 (T24) and 48 (T48) hours, and in the control group (untreated cells). The average viability ratios (n = 3) of cells and the relative standard errors were estimated through a Trypan blue dye exclusion assay and results were as follows: 62% ± 2.1 (T0, CM), 69% ± 2.3 (T0, SPF), 88% ± 2.2 (T24, SPF), 97% ± 0.5 (T48, SPF), and 98% ± 0.5 (control). CM and SPF T0 groups showed lower viability compared to the control group (p < 0.01). Conversely, no significant difference in viability was highlighted between the two treated groups (p > 0.05).
Primary cultured cells exhibited a uniform immunostaining with the fibroblast-specific marker vimentin (Fig. 1A), which confirms that they are a homogeneous cell population. Furthermore, they showed an elongated morphology and an in vitro cell growth typical of fibroblast cell population in both control (doubling time: 20–22 hours) and cryopreserved groups (doubling time: 22–24 hours; Fig. 1B).

Cryomicroscopy
Cryotests were performed in the presence of 10% Me2SO and without control of nucleation temperature (Tn). Both EIF and IIF were observed. IIF was assessed recording the exact temperature of cell darkening. In all samples, at least one cell underwent IIF together with an extracellular ice wave propagated inside the sample (Fig. 2); therefore, for each sample, the IIF temperature of the first darkened cell corresponded to the EIF temperature. Overall, each of the 14 samples analyzed (195 cells) showed a peculiar Tn, which ranged between −16.6°C and −29.3°C; however, a large part of the cells (45%) underwent IIF in a narrower range comprised between −20°C and −23°C, solely passing through the cooling phase (Fig. 2). Among the observed cells, 21% did not exhibit intracellular ice during the entire cryopreservation process at the cooling rate of −5°C/min (Fig. 3).
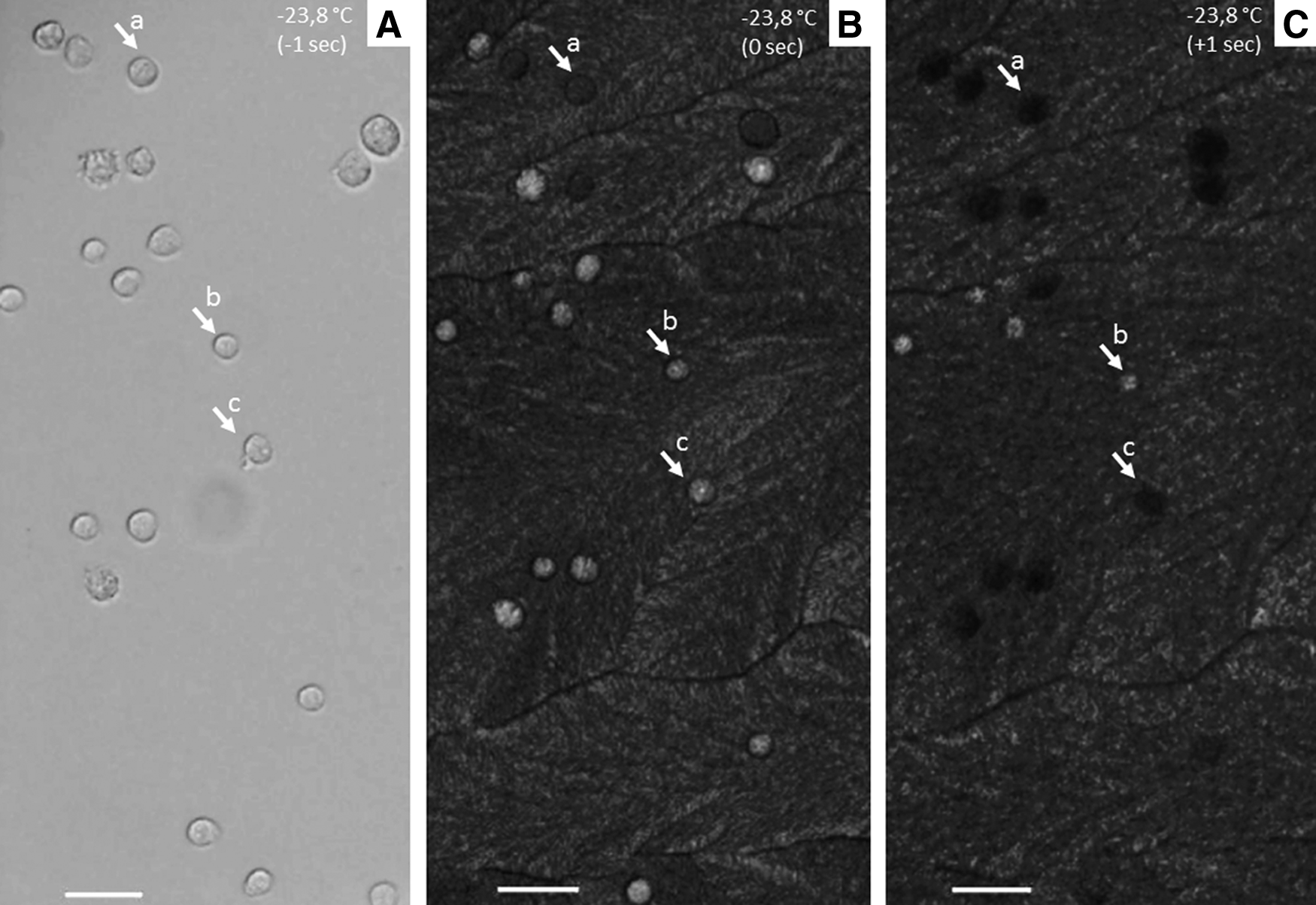
Cryomicroscopic view of fibroblast cells at −23.8°C during formation of extracellular and intracellular ice in cooling phase. Each frame was recorded at 1-second intervals.
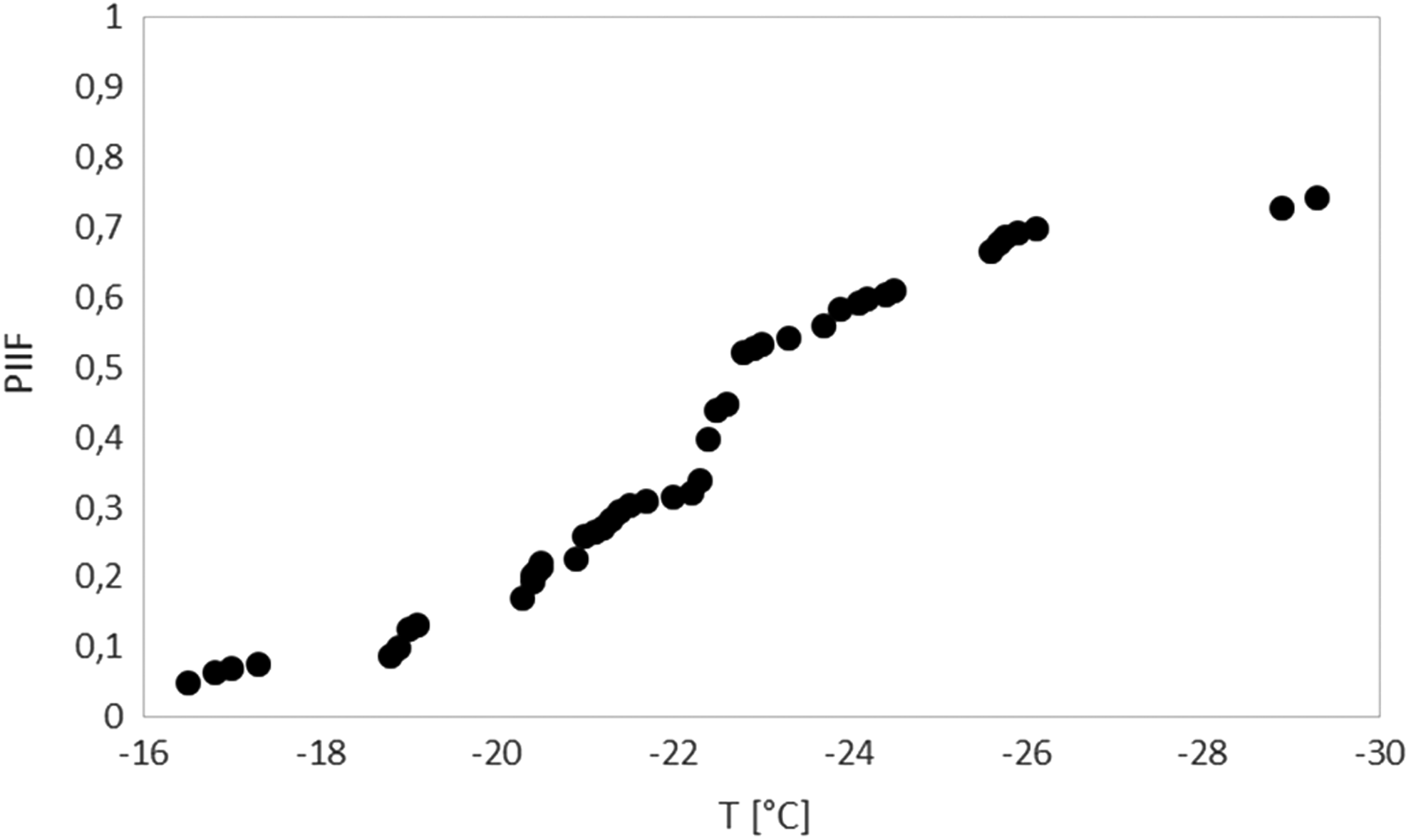
The plot shows the PIIF as a function of subzero temperatures based on cryomicroscopy videos recorded at 1 fps using a conventional CCD camera. The Y-axis shows the cumulative fraction of cells experiencing intracellular ice formation, which was visualized as darkening. Cell population of ovine fibroblasts was cooled at a rate of −5 K/min until −120°C and then at −150 K/min to the final temperature of −196°C. fps, Frame per second; PIIF, probability of intracellular ice formation.
The corresponding PIIF was determined as reported in Figure 3. We did not record any change in cell volume within the range of observation (+15°C/−20°C).
Gene expression analysis
The relative quantification of the transcripts is described in Figure 4. Cryopreserved cells display a higher presence of BCL2, OCT4, and GAPDH transcripts 24 hours post-thawing. Significant HSP90b and SOD1 upregulation was observed in cryopreserved cells at all time points. Conversely, no differences were observed in the quantity of BAX, CIRBP, or KIF11 transcripts between cryopreserved and control cells during the whole analyzed period.
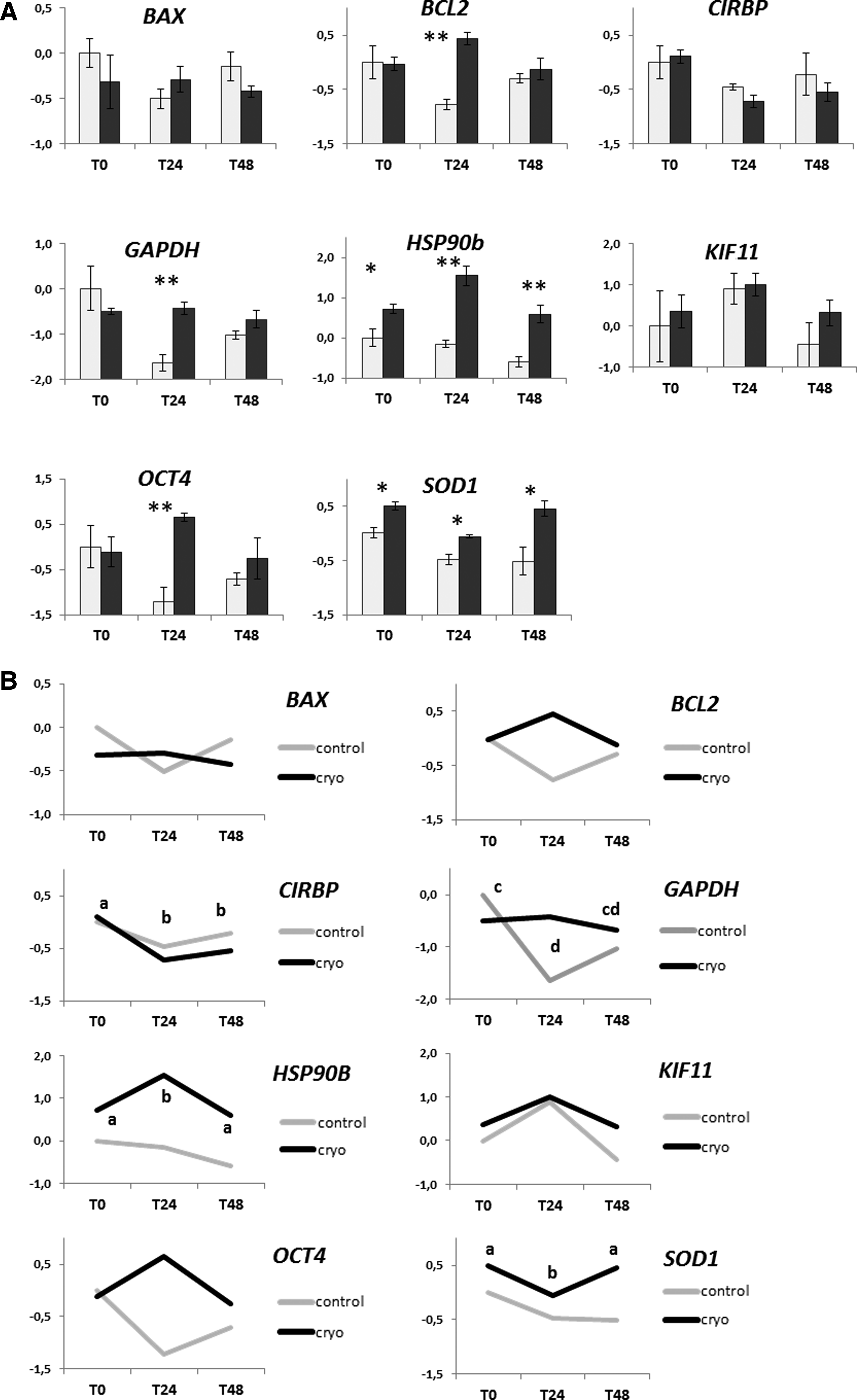
Relative expression of BAX, BCL2, CIRBP, GAPDH, HSP90b, KIF11, OCT4, and SOD1 in cryopreserved fibroblasts in vitro cultured for different time periods (T0 = analyzed immediately after thawing; T24 = in vitro cultured for 24 hours after thawing; T48 = in vitro cultured for 48 hours after thawing) and in relative controls not subjected to cryopreservation. Relative abundance values are expressed as ΔCq and show the mean value ± SEM of three replicates for each experimental group. Statistics were carried out using MINITAB software (release 12) using a general linear model ANOVA followed by Tukey's post hoc analysis. p values <0.05 were considered significant.
Discussion
Although cell cryopreservation is extensively used to preserve large batches of cells with unchanged functional properties, the alterations caused by cryopreservation procedures need to be carefully investigated. Membrane integrity assays, frequently used to assess cell viability, 6 are unable to give conclusive answers about the functional state of cells both during and after cryopreservation. This work described the effect of cryopreservation on CSSF examining dynamic IIF by cryomicroscopy and the expression status of a panel of genes after cell warming and subsequent 48 hours of in vitro culture.
Immediate or later alterations exerted by cryopreservation may limit the application of skin fibroblasts, especially when the cells are employed directly after thawing.
We observed that post-thaw (T0) cell viability in CM and SPF (62% vs. 69%) was not statistically different (p > 0.05) when a similar thermal profile was applied. More precisely, the two experiments differ mainly in the sample volume (5 μL vs. 210 μL) and in the thawing phase, that is, controlled versus uncontrolled warming rate, but share the same cooling rate. Even if this might potentially change the temperature for IIF when seeding is not used, the same post-thaw cell viability was obtained. Viability results are in line with those reported in previous studies that applied similar thermal profiles to cryopreserved cultured fibroblasts,2,4,12 while they are different from those reported by Choi and Bischof 7 and Leon-Quinto et al. 21 However, these studies were performed on other species and cells were cultured in different conditions. Moreover, we observed increasing values of cell viability in the SPF samples monitored through the 48 hours after thawing. This suggests that detrimental effects might require more time to manifest, as shown by the lower viability (69% T0 and 88% T24).
The CM allows direct observation of the events occurring inside the cell during the cooling and thawing processes. In particular, it is possible to identify the time and temperature when IIF takes place in a cell (i.e., when a blackening, flashing, or darkening cell is visualized). In this study, the results of IIF measurements are shown in Figure 3. The association of IIF with cellular blackening is still controversial. While Stott and Karlsson did not observe a strong correlation between darkening and primary IIF event, 30 several researchers detected darkening as a consequent phenomenon of IIF1,7,8,28,31,32,51 and used it to determine the PIIF.7,27 Along these lines, the results of the CM experiments reported in Figure 3 indicate that only 20% of the cells did not experience IIF. This is surprisingly lower than the 60%–70% post-thaw viability measured in terms of membrane integrity. It means that some cells are capable of maintaining membrane integrity in the presence of IIF. Since IIF is commonly associated with cell death in the field of cryobiology, our results suggest that sheepskin fibroblasts show some sort of recovery capability from the stress due to cryopreservation, at least in terms of membrane integrity.
Strictly related to IIF is the Tn, active control of which might improve the outcome of cell viability. 52 Although ice seeding is a recommended practice, several authors did not include an external nucleation step even with controlled freezing protocols.2,12–14,29,32,53–56 In our study, Tn was left to occur spontaneously to follow more closely the protocols applied during routine cell cryopreservation, where no seeding is performed (e.g., in vials or straws). Moreover, to properly record cell behavior throughout the entire length of the cryoprotocol, we avoided using the integrated seeding point of Linkam cryostage by moving the sample holder to the different physical position of the seeding post. In our experiments, without external seeding, the Tn ranged between −16.6°C and −29.3°C. The Tn was seen to vary also in human mesenchymal stem cells (hMSCs) cryopreserved in similar conditions. 32
Several studies have investigated the biochemical changes induced in somatic cells by cryopreservation procedures. Surprisingly, few studies have been performed in cryopreserved cultured fibroblasts; yet, such information could help evaluate the impact of cryopreservation on the main cell functions and improve their applications. As is already known, cell freezing triggers the expression of a set of genes involved in the recovery of cell damage, in apoptosis, and in the response to thermal stress.9,14–16,35,57 In a study focused on gene expression after cryopreservation, a selective upregulation of specific genes has been reported 48 hours post-thaw in cryopreserved thawed fibroblasts, with an increase in mRNA levels of VEGF, PDGF, and KGF growth factors and a decrease in transcripts encoding for matrix proteins. 13 In peripheral blood mononuclear cells (PBMCs), post-thaw gene levels can show an increased or decreased expression related to the cellular pathway where they are involved. 58 Moreover, the changes in gene expression may not occur immediately after thawing, but hours or days later. Baust et al. reported a delayed apoptotic response after cryopreservation that peaked 12–36 hours post-thawing, with significant rates of cell death often observed within 24–48 hours. 59 It has been reported that, for a reliable and consistent evaluation of cryopreservation effects, it is desirable to monitor cells at least during the first 24 hours following thawing.12,60,61 In agreement with this, our results showed that cryopreservation induces a significant increase in the expression levels of five out of the eight genes analyzed (Fig. 4). More specifically, we observed an increase in the expression of HSP90b and SOD1 genes in cryopreserved cells during the whole post-thawing culture (0–48 hours). The upregulation of the two genes is correlated with the cellular stress response and suggests an attempt by the cell to repair potential damage caused by cryopreservation. In fact, HSP90b is involved in the folding of other macromolecular structures, in redox state regulation, and in protein turnover, 35 while higher levels of SOD1 indicate oxidative stress determined by an increase in reactive oxygen species (ROS). 37
BCL2, GAPDH, and OCT4 genes showed a transient upregulation 24 hours post-thawing, which was no longer detectable 48 hours post-thawing, suggesting that cryopreservation caused an effect that the cells were able to control within a 48-hour timeframe. This was observed in BCL2, which codes for a protein involved in the antiapoptotic pathway, and suggests that cells may be able to overcome the barrier of the cell death mechanism, which might be triggered by temperature shock. Conversely, BAX, which is also a gene involved in apoptosis, was not affected by the procedure. A similar upregulation of BCL2 has been reported in cultured mesenchymal stem cells 3 hours after thawing, 38 in vitrified/thawed canine oocytes, 37 and in cryopreserved mouse embryos at the two-cell stage. 39
A transient increase in GAPDH expression was observed in cryopreserved cells 24 hours post-thawing. GAPDH was initially identified as a glycolytic enzyme and considered a housekeeping gene; however, its expression is actually tightly regulated and the enzyme is involved in numerous cellular functions. 62 Many of these roles are dependent on the ability of GAPDH to bind different macromolecules in the cell. Particularly intriguing are recent reports describing GAPDH as a regulator of cell death.40,41 The exact mechanisms by which GAPDH performs its nonglycolytic functions remain largely obscure; however, the variation in its mRNA and protein levels was observed in response to various stimuli42,63; indeed, the GAPDH gene and protein are also actively regulated on cell proliferation.64,65 This work includes cryopreservation in the range of stimuli that alter the enzyme expression.
Interestingly, we observed low levels of OCT4 mRNAs in in vitro-cultured CSSF and an unexpected increase in expression in cryopreserved fibroblasts 24 hours post-thaw. Oct4 is a key component of the pluripotency regulatory network (reviewed by Wu and Scholer 66 ); it is expressed specifically in the inner cell mass of the embryo and is involved in the maintenance of pluripotency in stem cells. 19 However, basal levels of OCT4 transcripts were reported also in somatic uncryopreserved cell lines, such as fibroblasts. 11 Notably, its expression was reported in response to different types of cellular stress such as heat shock 43 or cryopreservation. 44 Moreover, human fibroblasts cultured in a low-oxygen concentration environment showed a transitory increase in embryonic OCT4.67,68 In CSSF, the upregulation of OCT4 observed 24 hours post-thaw may be a rapid and transient reaction to the stress induced by cryopreservation due to the modified homeostatic conditions.
The dynamic variation in the expression of genes involved in the stress response, metabolic function, and cell viability indicated that these cells require at least 24–48 hours after cryopreservation to activate specific networks to reestablish their original physiological functions.
In conclusion, cultured sheep skin fibroblasts underwent relevant morphological and biochemical changes induced by cryopreservation procedures. These findings are relevant with a view to improving further applications of fibroblasts in several biotechnological fields (i.e., SCNT and iPS) where cryopreservation is a crucial step for proper storage of readily available stocks.
Footnotes
Acknowledgments
This work was supported by the “Regione Autonoma della Sardegna (RAS), legge 7—Promozione della ricerca scientifica e dell'innovazione in Sardegna and Progetti di ricerca fondamentale o di base annualità 2012—CUP F71J12000900002.” D.B. is the recipient of an RTD contract at the University of Sassari (Sassari, Italy), granted by “P.O.R. SARDEGNA F.S.E. 2007–2013—Obiettivo competitività regionale e occupazione, Asse IV Capitale umano, Linea di Attività l.3.1.” We thank Dr. Monica Strina for the proofreading and editing of the article.
Author Disclosure Statement
No conflicting financial interests exist.