
Abstract
Background:
The Legacy Biorepository is a College of American Pathologists-accredited biorepository operating within a seven-hospital healthcare system, with a decade's experience in specimen accrual, storage, and distribution. While standardization of our practices through accreditation remains a priority, we along with others face challenges with regard to sustainability. Purposeful changes in our consent process, which we term “progressive consent,” are expected to improve sustainability and operational flexibility while increasing our scientific impact.
Methods:
Until 2015, informed consent was performed primarily by biorepository staff at an estimated time of 1 hour per case. After a process improvement exercise, we successfully changed our informed consent process to a modified front-door model, with use of material and data for research as an opt-in or opt-out selection on the institutional patient informed consent form provided to surgery patients in the healthcare system. Successful implementation of this change required the engagement and participation of multiple stakeholders in healthcare system leadership, hospital administration, research, legal, regulatory, and patient care levels.
Results:
A modified front-door consent enabled us to collect an additional 38 specimens in the first two quarters of 2016, with a time commitment of 15.75 hours, a time savings per specimen increasing in Q2 over Q1. We estimate a potential savings of 43 hours in 2016. This progressive model allowed us to maintain our frozen sample collection while increasing the availability of paraffin-embedded tissue and bodily fluids. Augmenting our tissue collection added little expense per case (approximately half that of each frozen tissue aliquot) and increased the range of biospecimens collected.
Conclusions:
Biorepository financial sustainability is a critical issue. Thorough evaluation and modification of existing procedures and collection models, as well as cost recovery initiatives, can translate into savings. Sustainability, process improvement, and scientific impact broadly overlap and continue to require operational critique and implementation of strategic changes.
Introduction
B
Since its inception in 2006, the Legacy Biorepository has remained operational entirely due to philanthropic support from three local nonprofit organizations, two of which base their fundraising efforts for our program on our provision of specimens to investigators for shipping costs alone. To continue to grow the depth and diversity of our biorepository at a greater rate than the growth of our philanthropic support, we strategically worked to identify additional cost containment mechanisms. In addition, adding a formalin-fixed paraffin-embedded (FFPE) collection protocol to the biorepository became desirable for two reasons: (1) to capture previously formalin-fixed specimens and increase the diversity of our collection and (2) to retrieve specimens where fresh tissue was entirely utilized for diagnostic purposes. In 2014, we assessed the time we spent on processes, and analyzed workflows that required improvement and time savings. Our goal was to add new collection protocols with existing resources and to evaluate the sustainability of our workflows.
Protection of sample anonymity
We receive guidance for specimen utilization from a specimen use advisory committee and ethical oversight from our institutional review board (IRB). The samples maintained by our biorepository are characterized as deidentified specimens accessioned from remnant tissue collected during clinical care procedures. Therefore, the use of samples by recipient scientists does not constitute human subjects research, according to the prevailing interpretation. 3 While the biorepository maintains a link that identifies specimen donors, this information is withheld from recipient scientists. The identification link serves four purposes: (1) we are able to collect unanticipated data requested by recipient scientists, after treating these requests as retrospective chart reviews for IRB review; (2) we can collect longitudinal survival data; (3) retrospective data quality control assessments can be easily performed, for example, verification of pathologic characteristics and demographic data such as age and gender; and (4) donors may revoke their consent, that is, withdraw their existing donated samples from the biorepository.
All subjects (specimen donors) are enrolled according to informed consent requirements for human subjects research. Prior efforts to share the responsibility of conducting informed consent with healthcare providers yielded inconsistent results; thus, our own biorepository staff assumed the bulk of the responsibility for obtaining patient informed consent. The informed consent process requires a significant resource investment. The option of providing only deidentified samples to researchers and binding them under contract to make no attempt to identify the samples was discussed at length, to reduce the time and effort required to obtain donor consent. These discussions laid the groundwork for making changes beginning in 2015.
Addition of a paraffin-embedded tissue and bodily fluid collection protocol
Based upon our recipient scientist's requests, one of our goals was to initiate an FFPE tissue collection to include cases that were otherwise ineligible due to clinical care considerations. For example, snap-frozen tissue could not be provided from prostate cases due to diagnostic considerations; however, after FFPE cassettes were generated, the material availability for medical research could be assessed. Adding the FFPE collection would increase the number and types of materials available for collection; however, we did not have adequate resources to pursue the FFPE collection, while also remaining responsible for obtaining informed consent of all cases.
Cost assessment strategies
During the program development of the 2015 International Society for Biological and Environmental Repositories (ISBER) meeting, we were asked to test the Biobank Economic Modeling Tool (BEMT), developed by the United States National Cancer Institute's Biorepositories and Biospecimen Research Branch (US NIH/NCI BBRB). In 2011, our biorepository created novel operational cost calculators. Use of these cost assessment tools prompted us to examine workflows and job responsibilities in 2015, with one of the activities examined being the informed consent process. Based upon our internal assessments, the biorepository spent ∼60 minutes on consent per specimen accessioned (Table 1). The informed consent process impacts every specimen collected by the biorepository, which makes the potential for meaningful time savings strong. The consent process warranted attention based on the fact that it required the most time per sample, and is required for every sample collected. Our hypothesis was that time savings on informed consent would translate into meaningful cost reduction for the Legacy Biorepository.
Nearly 60 minutes was required to consent patients per specimen accessioned when using human subjects informed consent. The informed consent process impacts every specimen collected by the biorepository, which makes the potential for meaningful time savings strong when implementing front-door consent via our institution's standardized surgical consent form. We estimate a total savings of 43 hours in 2016.
Estimates based on total number of consents completed and estimated time commitment.
Estimates based on 38 cases collected from January 1 to June 30, 2016.
Efficiencies are improving and are expected to reduce the identification time to less than 5 minutes for 2017.
Regulatory and compliance issues
After discussions with our regulatory compliance officer, we jointly determined that the use of specimens collected by the biorepository qualifies under the prevailing interpretation of 45 CRF 46 as deidentified specimens, and their use in medical research does not qualify as human subjects research. In our biorepository, specimens collected by the biorepository and its donors are assigned codes that are not associated with any personal identifiers. In addition, recipient scientists are not provided with data that allow them to reasonably identify specimen donors. This includes specific and aggregate data. Finally, recipient scientists are required to sign a contract (material transfer agreement) that prevents them from attempting to identify or contact, or allowing a third party to identify or contact the donors of the material they receive from the Legacy Biorepository.
Other important factors include ethical issues and respect surrounding the donor. Our biorepository collects remnant tissue only, that has been removed from the patient for clinical care purposes, and which is not required for patient care. While use of the specimens does not meet the criteria of human subjects research, the fact that the specimens originate in human donors still requires their collection to be subject to review by an IRB. As mandated by the State of Oregon, patients are given the opportunity to “opt-out” of anonymous or coded genetic research (“anonymous” means that the patient could not be identified from the sample; “coded” means that the patient could only be identified where a separate code or encryption key is available to link the patient and the sample). While both anonymous and coded research make it very difficult for anyone to identify the patient source of the sample, the “opt-out” provision is an additional protection given to patients to ensure that their genetic information is not used for any purpose(s) beyond treatment, consistent with the patient's wishes. The Oregon law then allows patients to remain patients and not to become research “subjects” without their consent. 4
Practically, the advice to a patient regarding their right to “opt out” of the research is accomplished by a clear “notice” from the treatment provider of the patient's genetic privacy rights:
If a patient wants to allow their health information and biological sample to be available for anonymous or coded genetic research, they do not need notify Legacy. If a patient makes this choice, their health information or biological sample may be used for anonymous or coded genetic research without further notice to them. If a patient wants to decline to have their health information and biological sample available for anonymous or coded genetic research, they must tell their health care provider, clinic registration or patient access by requesting a ‘Notice of your Right to Decline Participation In Future Anonymous and/or Coded Genetic Research Genetic Opt-Out Form.’
5
A patient then can make an informed decision about whether to allow the legal “default” to remain in place or whether to “opt-out” of any kind of research on the leftover sample. Patients need only act proactively if they want to assert their genetic privacy rights by executing a simple form that is added to their medical record. If patients do not act proactively to exercise their right to “opt-out,” the provider notice informs them that their samples may indeed be used for future research and no further notice will be given to the patients. The samples can then be used. Most providers also include a statement on their notice that if the patients decide to opt out and refuse to allow their samples to be used, that decision will not affect their healthcare treatment or healthcare insurance coverage.
In our healthcare system, genetics material and information protection criteria are tracked in the electronic medical record. The genetics opt-out option is separate from the language included on our healthcare system's surgical consent form, which is only used for patients undergoing surgical procedures. Knowing if a patient had opted out of genetic research at the time of admission is an important consideration, since this would likely disqualify from participation in tumor banking.
We estimated that we could reduce the net minutes per sample collected through a streamlined consent process; namely, including an option on the standardized surgical consent form for the patients to donate their bodily tissue and/or fluid at the time of surgery (Fig. 1). This process only required the biorepository to track consent status and not approach patients for informed consent.

Revised Surgical Consent Tissue Donation Language. The figure is a snapshot of our institution's standardized consent form, which includes the section on donated tissue and/or bodily fluid.
We also determined that it was important to involve surgeons in the decision to make material available for biobanking at the front end of the treatment continuum. With the pathology department continuing to act as an “honest broker” for all potential donors, patient care would be protected from a diagnostic perspective over providing material for basic research.
Methods
In 2015, we proposed a global change in the surgical consent form template for our healthcare system and proposed this change as an IRB amendment. After obtaining IRB approval, the revision was vetted by the Legacy Health administrative leadership and legal department. Marketing and Community Affairs then helped to craft language that would not only conform to legal standards but also improve healthcare literacy by being intelligible to laypersons (Fig. 1). The new surgical consent form was reviewed by our healthcare system's Surgical Executive Committee before the approval by the Operations Council, the Executive Committee, and ultimately the Board of Trustees for subsequent implementation. This process took 9 months to complete and required additional time to educate surgeons and nursing staff on the added language and opt-in/opt-out option.
Between January 1, 2016 and June 30, 2016, we recorded the time required to complete consent verification for all surgical consent cases. As it has been our practice to designate time blocks to obtain consent, we recorded the total time required for each of eight steps of the consent process (Table 1). This time study was tracked for 38 specimens using the new progressive consent model.
Results
We determined that modified front-door consent enabled us to collect an additional 38 specimens in the first two quarters of 2016, with a consent process time commitment of 15.75 hours (25 minutes vs. 57 minutes per specimen, translating to a time savings of 32 minutes per specimen; Table 1). Per specimen time savings is expected to continue to improve as use of the new surgical consent form becomes more routine across all seven hospitals, as it has been implemented into the electronic format. Our revised estimate is a savings of 43 hours in 2016. In addition to the time savings, the numbers and types of samples collected have also diversified. Previous to implementing the surgical consent model, we focused exclusively on collecting snap-frozen tissue. The frozen collection protocol excluded certain types of tumors and had less representation of other types. Figure 2 shows the tissue region representation of the frozen collection, with additional FFPE specimens accrued during the 2015 calendar year. The addition of paraffin blocks has the potential to expand the types and numbers of samples collected. However, actual gains in sample types have not been realized at the time of publication. Additional time may generate changes to tissue region representation in the FFPE collection. Figure 3 demonstrates the proportional representation of tissue sites from the combination of frozen and FFPE collections between January 1, 2016 and May 31, 2016.

Frozen and FFPE Collections Tissue Sites, 2007–2016. This pie chart represents the diversity of our frozen collection, with the addition of paraffin-embedded specimens during the 2015 calendar year. FFPE, formalin-fixed paraffin embedded.
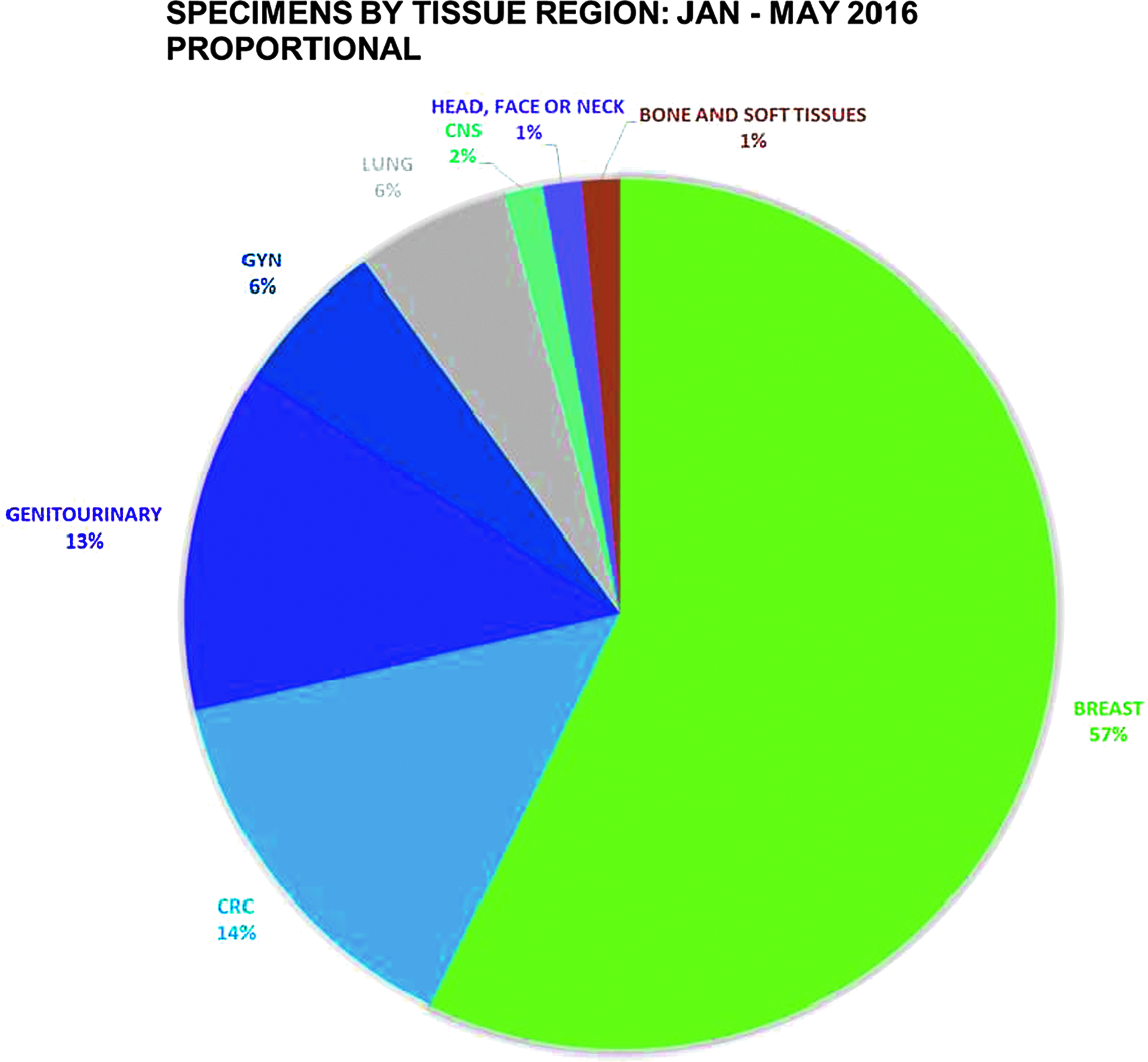
Frozen and FFPE Collections Tissue Sites, January 1, 2016 through May 31, 2016. This pie chart demonstrates the distribution of tissue sites from the combination of frozen and FFPE collections between January 1, 2016 and May 31, 2016.
Discussion
There were multiple challenges to the approval and implementation of revised language to the surgical consent form that were nevertheless accompanied by tangible benefits. A deeper understanding of Legacy Biorepository operations was necessary to continue support from institutional leadership. For example, communication with surgical leadership, research administration leadership, our healthcare system's Vice President and Chief Medical Officer, legal services, and our regulatory compliance department was required to make the argument for progressive consent compelling. In addition, discussion of changes to the surgical consent form also increased awareness of Legacy Biorepository activities among surgeons, managers, and surgical support staff system-wide. As a result of the new consent form, educational opportunities, primarily through in-service educational venues, increased general knowledge of Legacy Biorepository operations.
From an operational standpoint, opportunities were created to refine the protocol and educate surgeons on the Legacy Biorepository frozen collection protocol in the surgery suite. Education of nursing staff through annual in-service presentations not only helped in recruiting surgeons to consider helping to accrue donated specimens but also created the opportunity for positive discussion.
The complexity of our healthcare system's provider network required us to maintain two subject enrollment protocols after the tissue and bodily fluids donation language was successfully added to the surgical consent form template. This is because utilization of the new standardized consent form was initially made optional for some providers, who used prefilled consent forms and did not typically bank specimens. For this reason, we continue to maintain the human subjects research “long” consent form for potential use. The new protocol requires Legacy Biorepository personnel to review surgical consent forms for potential tissue and bodily fluid donation, and to check the status of each patient's genetics research opt-out form. The time commitment for any individual case is minimal (<5 minutes).
One limitation of this study is that the reported time savings is relatively small. However, we project that our progressive consent model will result in much high specimen accrual numbers than when we used our historical consent model. A longer follow-up period is required to determine the rate of increase in specimen numbers, and any associated time savings as a result.
From a sustainability standpoint, our biorepository operations improved by moving away from a traditional human subjects research consent process to a briefer request for permission to use deidentified specimens in multiple ways. We benefit from time savings and a modest expansion of our collection. Our gains were realized without compromising our continued respect for and ethical treatment of potential tissue donors. Our requirement for permission to use deidentified specimens protects patients' confidentiality from a legal standpoint, yet, also respects potential tissue donors' wishes regarding whether their bodily tissue and/or fluids are used for beyond what is required for diagnosis.
Conclusions
As the biorepository industry matures, financial sustainability becomes increasingly critical. Application of lean process improvement to evaluate and modify existing procedures and collection models can translate into savings and new cost recovery. Evaluation of workflows can help to identify areas where sustainability, process improvement, and scientific impact overlap; implementation of strategic changes that target these areas of overlap can result in improvements in all three areas.
Footnotes
Acknowledgments
The Legacy Biorepository wishes to acknowledge the Legacy Good Samaritan Foundation, the Treva Hoffman Foundation, and The Moto District for their generous financial contributions to our operational budget. In addition, we thank Cascade Pathology and Legacy Health for directing specimen contributions to our biorepository.
Author Disclosure Statement
No conflicting financial interests exist.