
Abstract
Skin vitrification is a promising and alternative tool for the conservation of biodiversity, especially for wild mammals, such as collared peccaries. Several factors can affect the success of this procedure, such as the cryoprotectant solution used. Therefore, this study was carried out to compare the efficiency of various vitrification solutions for recovery of viable cells after in vitro culture of cryopreserved skin tissues derived from the collared peccary, aiming to study the application in biobanking, where cellular use is not immediately required. Then, Dulbecco's modified Eagle's medium (DMEM) composed of 2.2 g/L sodium bicarbonate and 10% fetal bovine serum (FBS) was supplemented with 3.0 M ethylene glycol (EG) or 3.0 M dimethyl sulfoxide (DMSO) or 1.5 M EG plus 1.5 M DMSO with or without sucrose (SUC; 0.25 M) to produce six solutions for solid-surface vitrification. After warming, skin tissues were cultured in vitro and recovered cells were analyzed for morphology, adhesion, subconfluence, and proliferative activity for developing the growth curve and determining the population doubling time (PDT), and viability by Trypan Blue. The vitrification did not alter the ability of the tissues to adhere to the culture dish, as well as the day of all explants with cell growth, subconfluence samples, subconfluence total time, and PDT (p > 0.05). Moreover, independent of the cryoprotectant solution used, the vitrification altered the day of all attached explants (p < 0.05). Nevertheless, for viability after the first passage, only the EG-SUC (86.9%) and DMSO-SUC (91.4%) groups maintained viable cell recovery similar to the nonvitrified group (96.3%, p > 0.05). Additionally, for viability after the third passage, only the EG-SUC group maintained the cell quality (88.3%), when compared with the nonvitrified (97.8%, p > 0.05). In conclusion, DMEM with 10% FBS, 3.0 M EG, and 0.25 M sucrose was the most efficient solution for vitrifying collared peccary skin tissues, leading to the in vitro culture of viable cells.
Introduction
T
In this sense, somatic tissues derived from skin can be more easily obtained when compared with gametes and embryos 5 and tissue conservation by cryobanking can be permitted, maintaining a maximum representation for the population biodiversity. 6 Advances in cellular biotechnology increased the interest in the creation of these banks, especially with the possibility of reintroduction of lost genes through somatic cell nuclear transfer. 7 In 2009, 8 the first work in cloning an extinct animal was published, in which the authors produced the first clone of Capra pyrenaica pyrenaica, an animal derived from an extinct caprine subspecies using somatic cells as the donor of nuclei. Thus, the use of somatic banks would be interesting to allow an alternative way to produce gametes in endangered mammals. The development of suitable protocols for the preservation of skin tissues is of great interest, especially when the cellular use is not immediately needed. 1
Among cryopreservation methods, vitrification has been prominent for the conservation of somatic tissues.9,10 This procedure is characterized by rapid cooling essential to decrease the probability of ice crystal nucleation. 11 Among the techniques of vitrification used for skin samples in collared peccaries, Borges et al. 12 verified that solid-surface vitrification (SSV) is more suitable for tissue preservation when compared with direct vitrification in cryovials. Nevertheless, many factors still must be clarified for the success of tissue cryopreservation, such as the cryoprotectants used.
In general, cryoinjuries are caused by the formation of extracellular ice crystals and by the large amount of intracellular solutes that can cause cell death by necrosis or apoptosis. 13 These damages can be avoided by the proper use of cryoprotectants. On the other hand, the toxicity of cryoprotectants is the main barrier for the success of cryopreservation 14 and differences can be observed between species and cell types. 15 Moreover, cell types have different compositions in the phospholipid membrane, which affects the temperature transposition during cryopreservation. 7 The cryoprotectants most frequently used for tissue vitrification are ethylene glycol (EG) and dimethyl sulfoxide (DMSO), in association, or not, with sugars and proteins.16,17 The establishment of type and concentration of cryoprotectants for somatic tissue vitrification in collared peccaries is still needed.
In a previous study, 18 we observed by quantitative histology that EG with sucrose resulted in a better conservation of skin tissues derived from collared peccaries; nevertheless, the effect of these cryopreservation solutions on cell recovery after in vitro culture has not yet been observed in the species. Thus, the aim of the current study was to compare the efficiency of various vitrification solutions for recovery of viable cells after in vitro culture of cryopreserved skin tissues derived from collared peccaries.
Materials and Methods
This study was approved by the Ethics Committee of Animal Use of the Federal Rural University of Semi-Arid (CEUA/UFERSA; no. 23091.001072/2015-92) and the Chico Mendes Institute for Biodiversity Conservation (ICMBio; no. 48633-2).
Chemicals and media
All chemicals used were from Sigma-Aldrich (St. Louis, MO). Dulbecco's modified Eagle's medium (DMEM) and fetal bovine serum (FBS) were obtained from Gibco-BRL (Carlsbad, CA). Media were filtered using a 0.22 μm system (Corning, NY) and media pH was adjusted to 7.2–7.4
Skin biopsy and experimental design
Ear margin tissues (1–2 cm2) from five collared peccaries (3–6 months) were obtained from the Center for Wild Animals Multiplication (CEMAS/UFERSA, no. 1478912) and transported to the laboratory in DMEM supplemented with 2.2 g/L sodium bicarbonate and 2% antibiotic–antimycotic solution (penicillin G, streptomycin, and amphotericin B) at 37°C for 30 minutes. In management systems of this species, their identification has been recorded by ear sections, and these fragments were used for the experiments. In the laboratory, small tissue fragments (9.0 mm3) were washed in 70% ethanol and DMEM.
Then, tissue fragments were divided among nonvitrified (control) and vitrified groups. The vitrified fragments were prepared using DMEM composed of 2.2 g/L sodium bicarbonate, 10% FBS, and supplemented with EG, DMSO with or without sucrose (SUC; 0.25 M), as follows: EG (3.0 M EG), EG-SUC (3.0 M EG and 0.25 M sucrose), DMSO (3.0 M DMSO), DMSO-SUC (3.0 M DMSO and 0.25 M sucrose), EG-DMSO (1.5 M EG plus 1.5 M DMSO), and EG-DMSO-SUC (1.5 M EG plus 1.5 M DMSO and 0.25 M sucrose).
Skin vitrification and warming
Skin tissues were randomly allocated for each group and cryopreserved using SSV, according to Borges et al. 12 Briefly, fragments were exposed to 1.8 mL cryoprotectant solution for 5 minutes. Tissues were dried on absorbent paper, placed on a metal cubic surface partially in liquid nitrogen (LN2), transferred to cryovials, and stored in LN2.
After 2 weeks, for the warming process, cryovials were maintained for 1 minute at 25°C and immersed in a water bath (37°C) for 30 seconds. For removal of cryoprotectants, all fragments were washed three times for 5 minutes in DMEM with SUC in decreasing concentration (0.50, 0.25, and 0.0 M).
Primary culture and subculture preparations
Nonvitrified and vitrified fragments were seeded in culture dishes containing DMEM with 2.2 g/L sodium bicarbonate, 2% antibiotic–antimycotic solution, and 10% FBS, at 38.5°C, 5% CO2, and 95% air. The culture medium was changed daily. The cells were harvested when they reached 70% subconfluence, and were subcultured into dishes for 50 days of culture. 12 The 70% subconfluence was defined when 70% of the Petri dishes showed somatic cells. 19
Assessment of culture quality and proliferative activity
The daily evaluation of the cell culture quality from the onset until the subconfluence was performed under an inverted microscope (Nikon TS100, Tokyo, Japan) for the parameters: morphology, number of attached samples, number of samples with growth and subconfluence, day of attached samples, day of subconfluence samples, total subconfluence time, and adhesion ability of fragments.
The proliferative activity was quantified according to the determination of population doubling time (PDT) and development of the growth curve. Then, cells were seeded in 24-well plates (1.0 × 105 cells/well), 2 wells for each time per animal/group, cultured for 7 days, and counted daily to determine the number of cells. Data on cell growth and density were monitored and recorded, mean values were used to plot a growth curve, and PDT was calculated based on the growth curve.
20
Cell viability
Cell viability was determined by the Trypan Blue exclusion assay. Cells obtained from the first and third passage were suspended in culture medium; a cell aliquot was stained with 0.4% Trypan Blue for 2 minutes at 25°C and counted in the hemocytometer. 19 All evaluations were done in duplicate per animal/group.
Statistical analysis
All results expressed as mean ± standard deviation. Five repetitions were performed for each group, for one repetition/one animal. All values were verified for normality by the Shapiro–Wilk test and homoscedasticity by Levene's test. Data regarding the Trypan Blue assay did not show a normal distribution and they were arcsine transformed. All values were subjected to the variance analysis (ANOVA) followed by a paired and unpaired t-test using Stat View 5.0 (SAS Institute, Inc., Cary, NY) and considering significance as a value of p < 0.05.
Results
The vitrification did not alter the ability of the tissues to attach to the Petri dish, or the number of days of explants with respect to cell growth, subconfluence samples, and subconfluence total time (p > 0.05; Table 1). Moreover, regardless of the cryoprotectant solution used, the vitrification altered the day of all attached explants (p < 0.05; Table 1).
Within columns, values with different superscripts (a, b) are significantly (p < 0.05) different.
Five repetitions were performed for each cryopreservation solution, being one repetition/one animal.
Each animal resulted in 28 fragments distributed equally in 7 groups, resulting in 20 fragments of all animals per group.
Percentage calculated from initial number of skin fragments.
DMSO, dimethyl sulfoxide; EG, ethylene glycol; SD, standard deviation; SUC, sucrose.
The growth curve of somatic cells after vitrification (Fig. 1) appeared as a nontypical “S” shape and in all groups cells showed similar growth patterns for 7 days. Likewise, cells were characterized as fibroblast-like cells in the margins of tissue fragments. In general, all cells showed typical fusiform morphology, elongated with centrally located oval nuclei and active proliferation. Moreover, only the EG group showed a cell concentration lower than control (p < 0.05) for most days of the growth curve (Fig. 1). Additionally, the PDT was not altered by vitrification, which was ∼15.9 hours for cells derived from nonvitrified fragments, and 11.9–23.3 hours for cells derived from vitrified fragments.
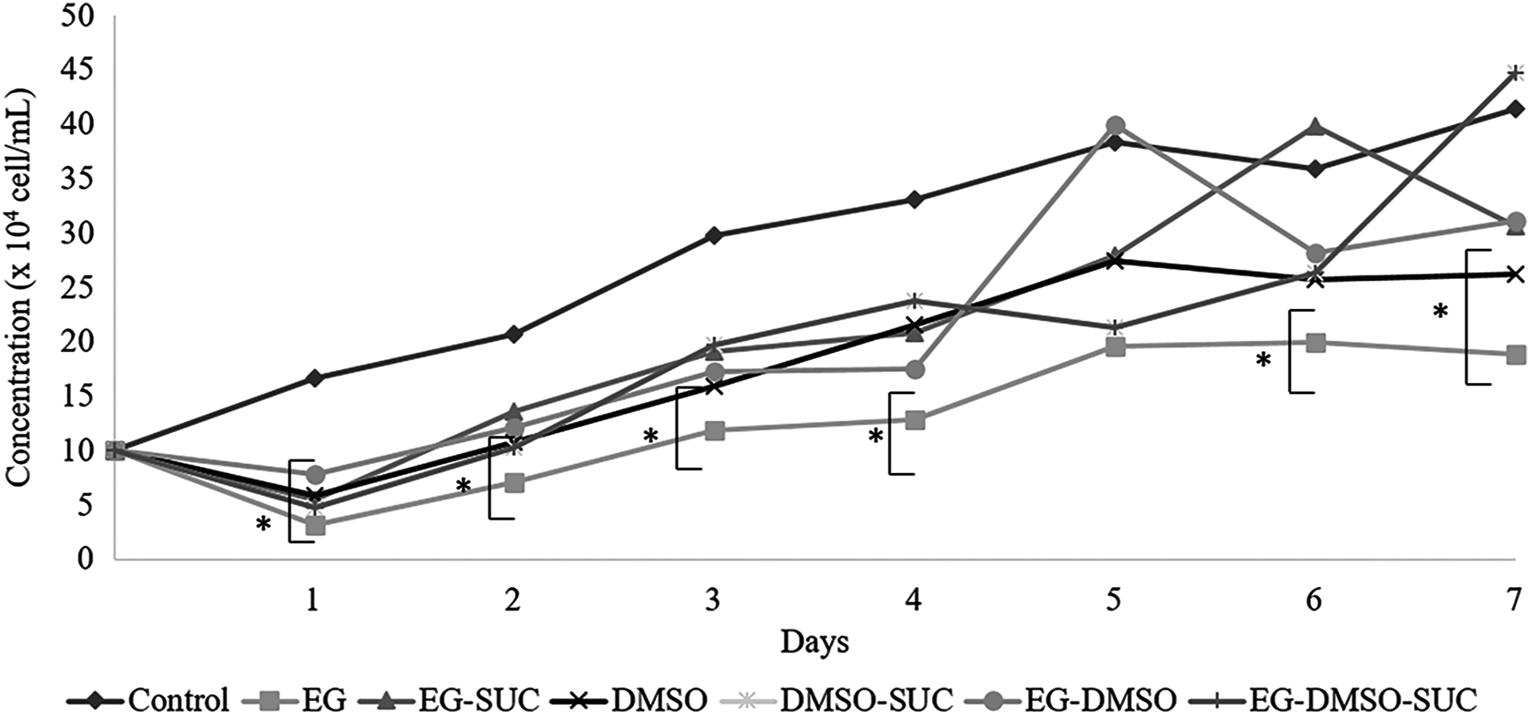
The growth curve of cells derived from skin tissues of collared peccaries in nonvitrified and vitrified fragments using different cryopreservation solutions. *Differences statistically significant for same time with the cells from control (p < 0.05).
Nevertheless, for viability after the first passage (Fig. 2A), only the EG-SUC (86.9%) and DMSO-SUC (91.4%) groups maintained viable cell recovery similar to the nonvitrified (96.3%, p > 0.05) group. Additionally, for viability after the third passage (Fig. 2B), only the EG-SUC group maintained the quality of the recovered cells (88.3%), when compared with the nonvitrified group (97.8%, p > 0.05).

The percentage of cell viability (mean and SD) after first
Discussion
In this study, we demonstrated the positive effect of combined sucrose to EG on the recovery of viable cells after in vitro culture of vitrified skin tissues of collared peccaries. The viability assay showed that EG-SUC vitrification solution was able to maintain cell membrane integrity during the vitrification process. The search for suitable protocols of cryopreservation is based on the central premise of maintaining cell integrity. 10 In this sense, the cell viability is an important parameter to indicate the protocol efficiency for the recovery of viable cells, and will allow the application of these cells for various purposes.7,21
These results corroborate those of Fahmy et al. 22 that demonstrated a low toxicity of EG on human cartilage tissue after chondrocyte recovery during in vitro culture. In general, the EG acts by penetrating the cell and osmotically replacing the intracellular water, preventing the formation of ice crystals, balancing intracellular proteins, and preventing membrane rupture. 23 Also, to reduce the hyperosmotic effect during cryopreservation, nonpenetrating substances are used, with sugars that act by dehydrating the cell, avoiding excessive turgidity and osmotic shock. 24
Moreover, our results corroborated the positive effect of sugars on the cell viability, for which they act as an important component of osmotic buffers, contributing to the general properties of vitrification. 25 Additionally, other tissues in different species also showed a positive influence of sucrose on the cryopreservation, such as for goat ovarian tissue, 26 equine ovarian tissue, 16 and human fetal skin. 24
In general, the ideal cooling rate also minimizes the formation of ice crystals. 27 In this sense, the SSV was able to promote an excellent cooling rate, which promoted, along with cryoprotectants, an adequate system of cryopreservation. Likewise, the presence of FBS in all vitrification solutions allowed the cell proliferation during in vitro culture, as well as the increase of the solution viscosity at low temperatures. 28 Additionally, proteins present in the FBS act as additives that favor the elasticity of the membrane, reducing cryoinjuires. 23
In summary, SSV using DMEM with 10% FBS, 3.0 M EG, and 0.25 M sucrose was the most efficient solution for vitrifying collared peccary skin tissues, allowing the stabilization of cell membranes and the inhibition of the formation of ice crystals during the skin vitrification. Therefore, the results generated to clarify the cryopreservation of somatic tissues increases the possibility of success for the formation of a sample germplasm bank.
Footnotes
Acknowledgments
This research was supported by the National Council of Technological and Scientific Development (CNPq) and Coordination for the Improvement of Higher Education Personnel (CAPES). The authors thank the CEMAS/UFERSA for providing the animals.
Author Disclosure Statement
No conflicting financial interests exist.