
Abstract
A major concern in biomedical research is the quality of biological samples. RNAlater is a stabilizer, which was originally developed for RNA preservation in fresh tissues and is important for collection and transportation. However, this reagent lacks a comprehensive and systematic evaluation of its preservative effect on different mammalian tissues under consistent experimental conditions. In this study, we collected liver, kidney, testis, brain, and colon tissues from mice and divided the samples into the following respective groups: fresh, RNAlater preserved, and liquid nitrogen snap frozen. Biomolecules (RNA, DNA, and protein) were extracted from each tissue in each group, and samples were formalin fixed and paraffin embedded for quality assessment. Our results revealed that high-quality (yield, purity, and integrity) nucleic acids could be extracted from all samples. Gene expression determined by quantitative real-time polymerase chain reaction exhibited no major difference among the three groups. Notably, we observed significant protein degradation in brain tissue preserved by RNAlater compared with fresh and snap-frozen tissue. Protein expression of the other four tissues was similar among the three groups. Hematoxylin and eosin staining of all tissue types indicated no apparent difference among the three groups. We concluded that high-quality nucleic acids can be obtained and tissue morphology conserved when tissues are preserved with RNAlater. However, there are tissue-specific differences in protein preservation when using RNAlater, which should be evaluated before extensive storage.
Introduction
T
RNAlater Stabilization Solution (Ambion, Darmstadt, Germany) is an aqueous ammonium sulfate tissue storage reagent. It was originally developed to stabilize and extract high-quality intact RNA in fresh samples by rapidly permeating tissues and deactivating nucleases. The application of RNAlater eased the collection and transportation processes for tissue samples, eliminating the need for LN or freezers on-site. 8 In addition, previous studies have displayed that high-quality RNA and DNA can be readily obtained from various types of RNAlater-preserved tissues from both humans and animals.9–13
Although previous research has indicated that high-quality nucleic acids can be obtained from RNAlater-preserved tissues,9–13 there are discrepancies regarding how this preservation affects protein quality and tissue morphology. In a previous study, Abbaraju et al. demonstrated that gulf killifish gill tissue proteins were better preserved by RNAlater, whereas the liver and heart tissues were preserved better by LN. 14 This suggested that RNAlater preservation has a different effect on proteins from different tissues in gulf killifish. Most existing research regarding mammalian animals only assessed a single tissue type and there were experimental differences among the different studies.15,16 Thus, there is insufficient research evaluating the protein preservative effect of RNAlater on different types of mammalian tissue.
Similarly, it is unclear whether RNAlater disrupts the morphology of tissues during the permeation process. Jhavar et al. obtained high-quality hematoxylin and eosin (HE) staining results from human prostate tissue preserved by RNAlater, 17 whereas another research group obtained poor results from RNAlater-preserved human ovary tissue. 18 This suggests that there are inconsistent results regarding the impact of RNAlater on the preservation of tissue morphology.
In light of these facts, comprehensive and systematic studies need to be conducted to evaluate the preservative effect of RNAlater on different tissues under consistent experimental conditions. In this study, we collected five commonly used types of tissues from healthy mice. Each tissue type was subsequently divided into three groups, including fresh tissues (F; as a standard control), RNAlater-preserved tissues (RL), and tissue snap frozen in LN. The preservative effects were determined by assessing the quality of extracted RNA, DNA, protein, and morphological changes of the tissue samples.
Materials and Methods
Animals and tissue samples preparation
Healthy 6-week-old male C57BL/6J mice were selected for tissue collection. Animals were used experimentally and permission was obtained from the ethical committee of Shanghai Children's Hospital. The animals were treated humanely and the standards conformed to those of current ethical animal research practices.
Mice were euthanized by diethyl ether and the heart was perfused with saline before dissection. Tissues from five different organs, including liver, kidney, testis, brain, and colon, were collected and cut into three sections on ice. The whole process was completed within 30 minutes.
The three respective sections of each tissue type were assigned to three groups: (1) the fresh group (F), where samples were collected with no pretreatments; (2) the RNAlater group (RL), in which samples were submerged in RNAlater overnight at 4°C and transferred to −80°C freezers for 6 days; and (3) the LN group, where samples were snap frozen in LN and stored for 1 week.
Biomolecule (RNA, DNA, and protein) extraction, formalin fixation, and paraffin embedding were performed immediately after collection for the fresh tissues. RNAlater- and LN-preserved tissues were removed from storage and handled by the same process as fresh tissues.
RNA extraction and quality assessments
RNA extraction
Preweighed samples of ∼15 mg each were submerged in 1 mL of Trizol reagent (Invitrogen, San Diego, CA). Tissues were homogenized with a mechanical homogenizer (Bertin Technologies, France), which was run for 10 seconds with a 20 second pause for six cycles. Subsequently, RNA was isolated per a standard phenol/chloroform RNA extraction procedure. In brief, the RNA was phase separated by the addition of 200 μL of chloroform and precipitated from the aqueous phase with 500 μL of isopropyl alcohol. Pelleted RNA was washed with 75% ethanol, dried, suspended in diethylpyrocarbonate-treated water, and stored at −80°C.
RNA yield, purity, and integrity
The quantity and purity of extracted RNA were evaluated using a NanoDrop™ 2000c spectrophotometer (Thermo Scientific, Waltham, MA) to determine RNA concentrations, as well as A260/280 and A260/230 ratios. RNA integrity was assessed by running 100–200 ng of sample in each lane of a 6000 Nano LabChip kit. Using an Agilent Bioanalyzer 2100 system (Agilent Technologies, Santa Clara, CA), we obtained the RNA integrity numbers (RINs) and 28S/18S ratios.
Reverse transcription
Reverse transcription was performed with a Prime Script™ RT Master Mix (Takara, Japan) kit. Complementary DNA (cDNA) was transcribed from 1 μg total RNA by adding 4 μL of 5 × Mix and RNase-free water to a total volume of 20 μL. The reaction mixture was incubated at 37°C for 15 minutes to perform reverse transcription and then 85°C for 5 seconds to inactivate the enzyme. The cDNA was stored at −20°C.
Quantitative real-time polymerase chain reaction
To assess the mRNA expression, quantitative real-time polymerase chain reaction (qRT-PCR) was performed on two genes (hypoxia-inducible factor 1 alpha subunit [Hif1a] and Finkel-Biskis-Jinkins osteosarcoma oncogene [Fos]) and respective tissue-specific genes (liver-specific Abcb11 and Slc10a1, kidney-specific Nphs1 and Nphs2, testis-specific Dazl and Ddx4, brain-specific NeuN and Gfap, and colon-specific Retnlb). Specific primers were designed for each gene (Sangon Biotech, Shanghai, China). The qRT-PCR reactions were carried out in a final volume of 10 μL, consisting of 1 μL of cDNA, 2 μL of nuclease-free water, 1 μL of forward primer (10 μM), 1 μL of reverse primer (10 μM), and 5 μL of 2× SYBR Supermix (Roche, Basel, Switzerland). The reaction was initiated for 10 minutes at 95°C followed by 45 cycles of 95°C for 10 seconds, and 60°C for 30 seconds. All reactions were performed on the Light Cycler96 PCR system (Roche). Expression levels were calculated from the mean cycle threshold (Ct) values for triplicates of each of the samples.
DNA extraction and quality assessments
DNA extraction
DNA was extracted from ∼10 mg of each tissue using a DNeasy® Blood & Tissue Kit (Qiagen, Venlo, the Netherlands). Samples were submerged in 180 μL of lysis buffer with 20 μL of proteinase K and homogenized using the procedure mentioned in RNA extraction. Per the manufacturer's instructions, total DNA was resuspended in Tris-ethylenediaminetetraacetic acid (Tris-EDTA) buffer and stored at −80°C.
DNA yield, purity, and integrity
DNA concentration was determined using a NanoDrop 2000c spectrophotometer (Thermo Scientific) and the purity was assessed by A260/280 and A260/230 ratios. Agarose gel electrophoresis (1%) was performed to evaluate DNA integrity.
Protein extraction and quality assessment
Protein extraction
Tissue samples of ∼20 mg each were submerged in 0.5 mL M-PER Mammalian Protein Extraction Reagent (Thermo Scientific) containing protease inhibitor cocktail. Tissues were then homogenized mechanically using the procedure described for RNA extraction. After centrifugation at 4°C for 15 minutes at 12,000 rpm, supernatant was collected and transferred to a new EP tube and stored at −80°C.
Western blots
Protein concentrations were determined using a Pierce™ BCA Protein Assay Kit (Thermo Scientific) and normalized with bovine serum albumin (BSA) as a standard. Absorbance at 590 nm was also determined using a Fluostar OPTIMA (BMG LABTECH, Ortenberg, Germany).
Western blots for HIF1A and FOS were performed on all samples from the three groups, whereas brain-specific NEUN was assessed on brain samples. Protein samples (10 μg) were denatured by incubation with loading buffer (Bio-Rad) and β-mercaptoethanol at 100°C for 10 minutes and then separated on sodium dodecyl sulfate–polyacrylamide gels (10%) and transferred to nitrocellulose membranes (YEASEN, Shanghai, China). After blocking in 5% nonfat milk in Tris-buffered saline with Tween-20 (TBS-T; Sigma Aldrich, St Louis, MO), the membranes were incubated with primary antibodies at 4°C overnight. The primary antibodies included mouse anti-HIF1A (1:2000 dilution; Abcam, Cambridge, United Kingdom), rabbit anti-FOS (1:2000 dilution; Abcam, Cambridge, United Kingdom), rabbit anti-NEUN (1:5000 dilution; Abcam), and mouse anti-ACTB (1:5000 dilution; Sungene Biotech, Tianjin, China). Membranes were washed with TBS-T and then incubated with the respective horseradish peroxidase-labeled secondary antibodies for 1 hour at room temperature. Goat anti-mouse and goat anti-rabbit (both 1:2500 dilution) secondary antibodies were used (Jackson Immuno Research, Suffolk, United Kingdom). Ultimately, chemiluminescent image analysis was performed using a ChemiScope 3500 (Clinx, Shanghai, China). Bands were quantified by ImageJ software.
HE staining
Tissues of all three groups were formalin fixed, paraffin embedded, and cut into 4-μm-thick sections. Sections were subsequently deparaffinized in xylene and absolute ethanol, then rehydrated by successive immersions in 95% ethanol, 70% ethanol, and distilled water. All sections were HE stained and observed by light microscopy to evaluate the morphological changes.
Statistical analysis
Statistical analysis was performed using SPSS Statistics 19.0 (IBM, Chicago, IL). The different nucleic acid yields, A260/280 and A260/230 ratios, RINs, 28S/18S ratios, gene expression, and protein quality data among the different groups were validated by the one-way analysis of variance. This was followed by a Tukey's post hoc pairwise comparison test. A p-value of <0.05 was considered to be statistically significant.
Results
High-quality nucleic acids were extracted from all five types of tissues preserved by RNAlater
In accordance with previous studies, RNA extracted from samples preserved by RNAlater exhibited high quality. In all studied tissue types, the RNA yields from the RNAlater-preserved tissues were similar to the fresh and snap-frozen samples (Fig. 1A). The purity, measured as A260/A280 and A260/A230 ratios ranging from 1.80 to 2.16, also exhibited no significant differences among the three groups (Fig. 1B, C). The RIN values were all >7 and the 28S/18S ratios ranged from 1.10 to 1.65 in the three groups, and were not significantly different (Fig. 1D, E). To determine whether RNAlater affected gene expression in tissue samples, qRT-PCR was performed on the mRNA. The genes Hif1a, Fos, and the tissue-specific genes were studied in all five tissue types. There was no major difference in the gene expression within the three groups (Fig. 2A–E).
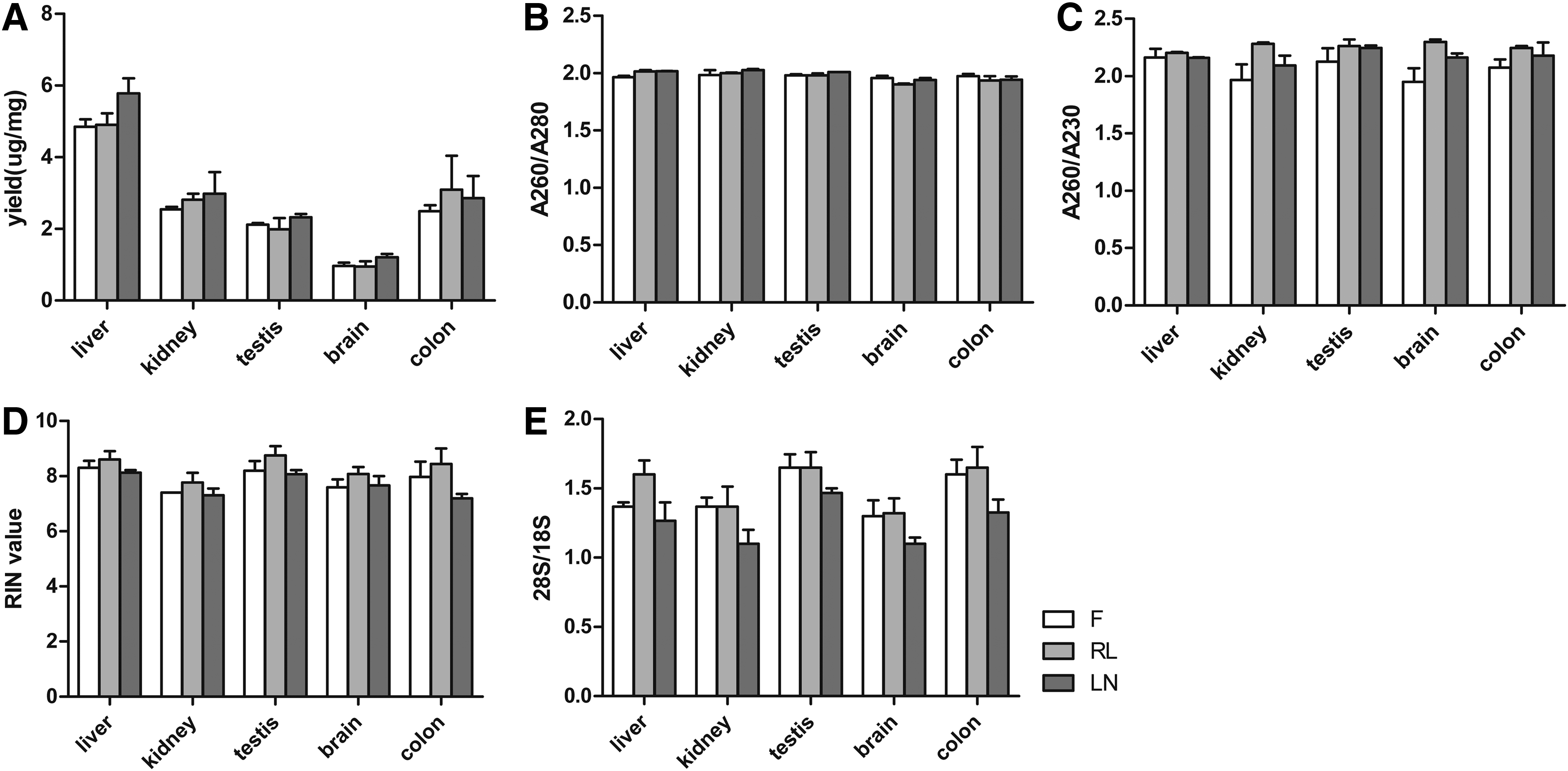
Assessment of RNA quality.

Gene expression of all tissues in the three groups analyzed by quantitative real-time polymerase chain reaction.
High-quality DNA was extracted from all samples. In comparing the three preservation methods, similar yields of DNA were obtained from each tissue type (Fig. 3A). A260/A280 and A260/A230 ratios ranging from 1.91 to 2.18 showed no significant difference in DNA purity among tissue samples from the three groups (Fig. 3B, C). Agarose gel electrophoresis of DNA from the preserved tissue samples showed no significant differences in DNA integrity between the three groups (Fig. 3D). Nucleic acids extracted from RNAlater-preserved tissues were not more degraded than fresh or snap-frozen tissues.

Assessment of DNA quality.
Tissue-specific protein degradation was observed in brain tissue preserved by RNAlater
Protein quality was evaluated by determining the expression of protein HIF1A and FOS using western blotting. Our results demonstrated tissue-specific effects on RNAlater-preserved protein samples. As indicated in Figure 4A, RNAlater-preserved liver, kidney, testis, and colon samples had similar protein preservation to the fresh and snap-frozen groups. However, protein HIF1A and FOS were degraded in brain tissues preserved by RNAlater, whereas no major difference was observed between fresh and snap-frozen samples (Fig. 4B). To demonstrate this phenomenon, the brain-specific protein NEUN was later analyzed by western blotting. Consistent with HIF1A and FOS, protein degradation of NEUN was also detected in the RNAlater-preserved sample (Fig. 4B).

Western blot analysis of protein expression in the five tissue types from the three preservation groups.
Tissue morphology was preserved in all five types of tissues preserved by RNAlater
To examine whether RNAlater preservation resulted in morphological changes to the tissue samples, we applied HE staining to all of the tissue types from the three groups. As indicated in Figure 5, the morphology of RNAlater-preserved tissues showed no apparent difference from fresh and snap-frozen tissues.
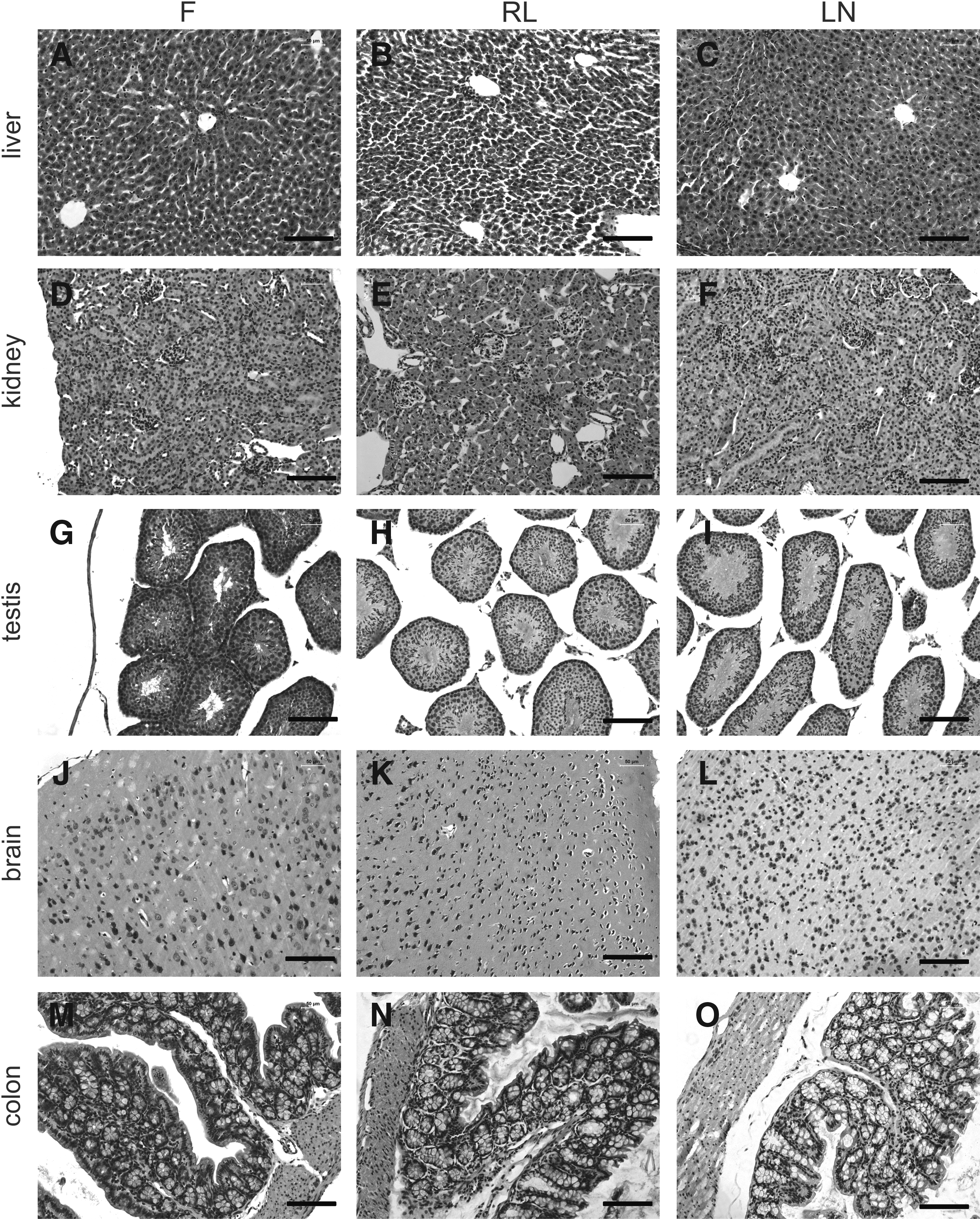
Morphology of tissue samples from the three groups.
Discussion
High-quality biological samples are vital for translational research and precision medicine. To maintain the long-term stability of tissue samples, LN or −80°C freezers are widely accepted.4–7 From acquisition to long-term storage, samples usually need to be transferred from operating rooms to biobanks or between institutes for multicenter research. Regarding safety and convenience, RNAlater Stabilization Solution offers a suitable alternative to traditionally used LN or dry ice for tissue transportation. However, as an additive into tissues, it is more likely to cause biological changes when compared with frozen methods without any additional compound. 16 As such, the effect and impact of RNAlater on different tissues remain poorly described.
In this study, we simultaneously examined the quality of RNA, DNA, protein, and the changes to tissue morphology in five commonly used mouse tissues under consistent experimental conditions. Our research indicated that RNAlater preservation provides high-quality nucleic acids and conserved tissue morphology from all five tissues. The quality and quantity of nucleic acids obtained from RNAlater-preserved tissues was not significantly different from fresh and snap-frozen tissues. However, there were tissue-specific preservative effects of RNAlater on protein in the brain samples, which differed significantly from the fresh and snap-frozen brain samples.
Nucleic acids extracted from tissues preserved by RNAlater exhibited high quality in this study, similar to those from fresh and snap-frozen tissues. As was seen in previous studies, RNA from various tissues and different species is well preserved by RNAlater and is suitable for RT-PCR, cDNA arrays, and next-generation sequencing.9–11,19–22 In this study, we evaluated the mRNA expression using qRT-PCR. A hypoxia-induced gene (Hif1a) and an immediate early gene (Fos) were studied in all five tissue types. They express in different types of tissues or cells, but the expression levels may be affected differently by the experimental conditions, such as surgical manipulation, warm ischemia, and processing before storage. 23 We also measured several tissue-specific genes but found no major difference in the mRNA expression in these different groups. Regarding DNA preservation, previous research has shown that high-quality DNA is obtainable from various tissues preserved by RNAlater.12,13 However, in a study conducted by Allen-Hall and McNevin on disaster victim identification in a tropical climate, storage conditions were observed to be important. 24 To simulate the natural environment, researchers stored samples at 35°C. They assessed several tissue preservatives and found that RNAlater-preserved human muscle tissues showed significant DNA degradation. Since enzyme activity is temperature sensitive, 25 we reasoned that the degradation resulted from the high storage temperature. 24 Taken together, we suggest RNAlater as a suitable tissue stabilizer for nucleic acids preservation when samples are stored at 4°C.
In this study, preservation of brain samples with RNAlater differed from liver, kidney, testis, and colon samples. Abbaraju et al. have demonstrated that RNAlater showed different preservative effects on protein from different gulf killifish tissues. 14 Nevertheless, there was insufficient evidence to confirm that phenomenon in mammals. According to previous research, high-quality proteins can be extracted from RNAlater-preserved mammalian tissues, such as human colon mucosal biopsies, 15 rat mesenteric arterial and venous tissue, 26 and mouse testis. 27 Conversely, Wang et al. detected protein degradation in RNAlater-pretreated human breast cancer tissue samples. 16 Not only were there methodological differences between these two studies, but also the earlier research was mainly performed on a single mammalian tissue type. In addition, these results could be biased by the experimental environment, multiple batches of biological reagents, different data processing methods, and many other technical variations. To overcome this bias, we performed histological experiments on different organs simultaneously. We found that in murine tissue, protein degradation occurred in RNAlater-preserved brain tissue. This degradation was not observed in the liver, kidney, testis, and colon samples from the three groups we tested, where no difference was observed. We speculate that this may be due to tissue-specific differences in cell structure, intracellular metabolism, or enzymatic functions. This will be investigated in our future studies. Meanwhile, it is important to note there are tissue-specific differences on protein preservation when using RNAlater. A trial assessing storage and preservative effects should be conducted for specific tissue types before extensive storage of biological samples. This is especially important when it is unclear which experimental methods will be performed in subsequent analysis.
Our findings in tissue histological analysis indicated that tissues preserved by RNAlater maintained satisfactory morphology. These data were in accordance with previous studies involving human prostatic, skin, cerebellum, thyroid, gastric, and tongue tissues.17,28 However, a discrepancy exists between the earlier studies and the result of Staff et al., who found poor morphological HE staining resulting from RNAlater-fixed human ovary and endomyometrium sections. 18 The discrepancy probably results from tissue processing procedures, since this group used RNAlater as a fixative before dehydrating and paraffin embedding the tissue. 18 In our study and in previous research,17,28 RNAlater was used as a tissue stabilizer followed by formalin fixation, dehydration, and paraffin embedding. Therefore, we suggest that RNAlater preservation will not negatively impact tissue morphology, and can be used for HE staining and microscopy examination.
In conclusion, high-quality nucleic acids can be obtained and tissue morphology preserved when using RNAlater on various types of tissues. When LN or ultralow-temperature freezers are not available on-site, tissue samples can be submerged in RNAlater at 4°C overnight and then transferred to −80°C freezers. The quality of these tissues is suitable for subsequent nucleic acids and morphological analysis. However, there are still tissue-specific differences observed in RNAlater-preserved proteins. These tissue-specific effects should be evaluated for specific tissue types before extensive storage of biological samples.
Footnotes
Acknowledgments
This study was supported by funding from National Nature Science Foundation of China (81270742, 81370700, 81470911), funding from Shanghai Municipal Commission of Health and Family Planning (201540389), Professional and Technical Services Platform for Biobank of Critical Disease in Shanghai (15DZ2292100), and Interdisciplinary Funding of Medical and Engineering from Shanghai Jiao Tong University (YG2016MS32).
Author Disclosure Statement
No conflicting financial interests exist.