
Abstract
During pulmonary resections, removal of visceral pleura is frequently required, resulting in lung air leakage (LAL) and bleeding. Especially persistent LAL after pulmonary surgery has negative consequences. Current surgical procedures are ineffective in closing these visceral pleural injuries. Previously, the authors' laboratory has developed a novel and effective LAL sealant using tissue-engineered cell sheets harvested from temperature-responsive culture dishes. The clinical application of fresh fibroblast sheets (FSs) is limited by several problems related to the cell culture period, mass production, preservation, and transportation. Therefore, cryopreservation of FSs and feasibility of off-the-shelf FSs for repairing visceral pleural defects were investigated. Over 3 to 6 months, harvested skin-derived FSs in Dulbecco's modified Eagle's medium supplemented with 10% dimethyl sulfoxide were stored in an atmosphere of liquid nitrogen. The amounts of cytokines (basic fibroblast growth factor [bFGF] and vascular endothelial growth factor) released from frozen–thawed FSs were determined. bFGF levels were significantly elevated in frozen–thawed FSs compared with fresh FSs. After a visceral pleural injury model was created, a frozen–thawed skin-derived FS was transplanted directly to the defect. One month after transplantation, the frozen–thawed FS remained on the pleural surface, resulting in permanent closure, suggesting that cells in the off-the-shelf FS had the ability to proliferate and release various cytokines. Frozen–thawed FSs were useful for closing LALs during pulmonary surgery as an off-the-shelf technique and would be used as a pleural substitute.
Introduction
During pulmonary resections, removal of visceral pleura is frequently required, resulting in a lung air leakage (LAL) and bleeding. LAL that persists after pulmonary surgery has negative consequences on overall morbidity, hospitalization, and health care costs.1–6 While some LALs can be avoided with careful surgical techniques,2,5–7 many techniques previously advocated as LAL sealing methods can only promote healing and prevent fistulization. However, current surgical procedures are generally ineffective in closing these visceral pleural injuries.4–8 Previously, the authors' laboratory has reported the use of (1) temperature-responsive culture dishes (TRCDs) for preparing transplantable cell sheets comprising living cells with intact extracellular matrix (ECM) and (2) an effective cell–sheet LAL sealant.9–13 The results with skin-derived fibroblasts confirm that fresh fibroblast sheets (FSs) can be used as durable, long-term LAL sealants that can respond according to the dynamic movements of lungs during respiration and can adhere to tissue surfaces repeatedly, owing to their natural adhesive proteins. However, clinical application of fresh FSs is limited by problems related to the necessary cell culture period, mass production, preservation, and transportation. The biggest problem is that FSs are unavailable to be used for emergency surgery. Among cases in which pulmonary resection is performed, their primary pneumothorax and secondary pneumothorax often recur in a short period. At the time of initial or latest operation, fibroblasts are isolated from the patient's own excised skin, and FSs are fabricated and cryopreserved. In a clinical setting, cryopreserved FSs are expected to be used immediately at the onset of pneumothorax. There is no specific preservation method for cell sheets. Therefore, this study attempted to cryopreserve FSs and highlight the feasibility of off-the-shelf FSs for repairing visceral pleural defects.
Materials and Methods
A small piece of skin was obtained from a patient during surgical excision in compliance with the ethical guidelines of Tokyo Women's Medical University. Oral and written informed consents were obtained from individual patients. Patients were confirmed to be free from infections and viruses, including hepatitis B virus, hepatitis C virus, and human immunodeficiency virus, and also negative for the Treponema pallidum hemagglutination test.
All animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the National of Institutes of Health (Publication No. 85–23, revised 1996) and the Guidelines of Tokyo Women's Medical University on Animal Use.
Preparation of square-patterned, poly(N-isopropylacrylamide)-grafted cell culture dishes
The preparation of square-patterned TRCDs provided by CellSeed (Tokyo, Japan) has been previously described. 14 Briefly, N-isopropylacrylamide monomers in 2-propanol solution were spread onto 35-mm-diameter tissue culture polystyrene dishes. Dishes were then irradiated by an electron beam, resulting in both polymerization and covalent grafting of poly(N-isopropylacrylamide) (PIPAAm) onto the cell culture surfaces.
PIPAAm-grafted dishes were rinsed with cold distilled water to remove ungrafted monomers and dried in nitrogen gas. To prepare dishes having square-geometry temperature-responsive surfaces, PIPAAm-grafted surfaces were masked with square glass coverslips (24 × 24 mm; Matsunami, Osaka), and acrylamide (AAm) monomer (Wako Pure Chemicals, Osaka) in 2-propanol was spread onto the masked dish surface. Then, the dish surfaces were irradiated again in the same manner. The resulting culture dishes had center square areas grafted with temperature-responsive PIPAAm with the surrounding area of noncell-adhesive poly-AAm. All temperature-responsive culture surfaces were eventually gas-sterilized with ethylene oxide.
Culture media
Primary culture and subsequent passages were carried out in culture medium consisting of Dulbecco's modified Eagle's medium (DMEM) (Sigma, St. Louis, MO) supplemented with 10% fetal bovine serum (Moregate Biotech, Queensland, Australia), 100 U/mL penicillin and 100 μg/mL streptomycin (Gibco BRL Life Technologies, Grand Island, NY), and 2.5 μg/mL fungizone (Gibco).
Culture of human skin-derived fibroblasts
Skin specimens (leaf shape, size: 10 × 5 mm) were excised from the lateral chests of patients (three men and two women; mean age 79.2 ± 2.8 years) who were operated on with video-assisted thoracic surgery under general anesthesia, and the remaining skin incisions were used for minithoracotomy. The excised tissues were cut into 3 × 3-mm pieces and treated with DMEM containing 1000 U/mL dispase I (Godo Shusei, Tokyo) overnight at 4°C. After enzymatic treatment, all epithelial layers were removed from the underlying dermis by surgical forceps under a dissecting microscope. The dermis was then finely minced and subjected to dissociation with 0.05% collagenase (Wako Pure Chemicals) at 37°C for 10 minutes with gentle shaking. Enzymatically treated cell suspensions were then centrifuged and resuspended in fresh collagenase solution, and the process was repeated in the same manner three additional times. Skin-derived fibroblasts were then plated onto a commercially available 60-mm-diameter tissue culture dish (BD Biosciences, Franklin Lakes, NJ) at an initial density of 2.0 × 105 cells/cm2 after enzymatic treatment, cultured for 2 weeks at 37°C in a humidified atmosphere of 5% CO2, and the culture medium was changed every 2 days.
Primary cultured cells were subsequently harvested by treatment with 0.05% trypsin–ethylenediamine tetraacetic acid (EDTA) (Sigma) for 15 minutes at 37°C. The expanded cells were seeded on square-patterned TRCDs at a density of 1 × 105 cells/cm2 and cultured for one additional week. Cells cultured on temperature-responsive surfaces were then transferred to a separate incubator set at 20°C for noninvasive cell–sheet harvest.
Cryopreservation of harvested skin-derived FSs
FSs made from harvested skin fibroblasts were washed with phosphate-buffered saline (PBS) three times. Washed FSs and 1.5 mL of DMEM supplemented with 10% dimethyl sulfoxide (DMSO) (Sigma), as a cryoprotectant, were placed in 2-mL internal threaded polypropylene cryovials (Corning, NY), which were placed into a −70°C freezer overnight, transferred to an atmosphere of liquid nitrogen on the next day, and stored in liquid nitrogen for 3 to 6 months.
In vitro measurements of growth factor secretion
Levels of cytokines secreted from FSs were measured by enzyme-linked immunosorbent assay kits. Cryopreserved FSs were thawed by warming at 37°C. Frozen–thawed FSs were then rinsed with PBS to remove the cryoprotectant. Frozen–thawed FSs were incubated in serum-free DMEM to avoid overestimation of growth factor secretion. After a 24-hour incubation, amounts of basic fibroblast growth factor (bFGF), hepatocyte growth factor (HGF), and vascular endothelial growth factor (VEGF) in the media were determined, according to the manufacturer's suggested protocol. As the control, fresh FSs were incubated in serum-free DMEM. After a 24-hour incubation, culture media from each sample were then collected, and amounts of bFGF, HGF, and VEGF in the media were determined.
Cytotoxic evaluation and cell viability of frozen–thawed FSs
Both frozen–thawed and fresh FSs in serum-free medium were placed on 60-mm-diameter culture dishes (BD Biosciences) and incubated for 24 hours at 37°C in a humidified atmosphere of 5% CO2. The culture media from each sample were then collected. The amounts of lactate dehydrogenase (LDH) released from both frozen–thawed and fresh FSs were measured by the Japan Society of Clinical Chemistry transferable method. For evaluating cell viability in frozen–thawed FSs, the FSs were replated on 60-mm-diameter culture dishes (BD Biosciences) and incubated at 37°C in a humidified atmosphere of 5% CO2. After reculture, the status of fibroblasts in cell sheets was observed with a phase-contrast microscope (ECLIPSE TE2000-U) (Nikon, Tokyo).
Transplantation of off-the-shelf sheets to pleural injury
For frozen–thawed FS transplantation, five male F-344 athymic rats (8 weeks old) (Charles River Japan, Tokyo) were anesthetized with 2% inhaled isoflurane and ventilated by a rodent mechanical ventilator. In a sterile condition, the animals were positioned in a right lateral decubitus position, and a left lateral thoracotomy in the fifth intercostal space was performed. A 5-mm-long incision with a depth of 3 mm was made in the left lung by sterile surgical scissors. LAL was confirmed by the continuous appearance of air bubbles upon submergence in physiological saline after the initial injury. LAL was then closed with transplantation of the frozen–thawed FS. Five minutes after the closure, the surgically operated lung was again submerged in saline solution to verify the presence or absence of air bubbles.
All rats were not treated with drugs such as steroid and immunosuppressive agents after surgery. After 1 month, a left lateral thoracotomy in the fifth intercostal space was performed under anesthesia, as described above. The transplanted area was carefully inspected, and the left lung, including the surgically operated site, was subsequently resected for further analysis.
Histological analyses
Both FSs and tissues were fixed with 10% formalin and routinely processed into 10-μm-thick paraffin wax-embedded sections. Hematoxylin and eosin and Azan staining were performed by conventional methods.
Electron microscopy
Rat lungs were fixed with 2.5% glutaraldehyde and postfixed with 1% osmium tetroxide. The specimens were dehydrated through a graded series of ethanol and embedded with the Poly/Bed 812 embedding kit (PolyScience, Warrington, PA). Ultrathin sections were then stained with uranyl acetate and lead citrate and observed with a transmission electron microscope (Hitachi H-7100) (Hitachi, Ibaraki) at a magnification of 4000.
Data analysis
All data are expressed as mean ± standard deviation. An unpaired Student's t-test was performed for comparing two groups. A probability value <0.05 was considered to be significant.
Results
Fabrication of off-the-shelf FSs
Skin-derived fibroblasts were isolated from the lateral chest skin of a human and expanded before seeding on TRCDs. After a 2-week culture, fibroblasts proliferated to form confluent monolayers (Fig. 1A). To prepare skin-derived FSs, primary cultured cells were trypsinized and seeded onto square-patterned TRCDs. After an additional 1-week culture, intact 24 × 24-mm sheets of skin-derived fibroblasts were able to be harvested by simple temperature reduction to 20°C (Fig. 1B). The harvested skin-derived FSs consisted of one to three stratified cell-dense layers with relatively little ECM present (Fig. 1C). Elongated fibroblasts were surrounded by collagen and other connective tissue (Fig. 1D). Over 3 to 6 months, the harvested skin-derived FSs in DMEM supplemented with 10% DMSO were stored in an atmosphere of liquid nitrogen. The cryopreserved FSs were thawed by warming at 37°C (Fig. 1E). The frozen–thawed FSs consisted of one to three stratified cell-dense layers with ECM, and the cell density of the off-the-shelf FS decreased (Fig. 1F). In the transmission electron micrograph of the frozen–thawed FS, fibroblasts had vacuolated cytoplasm without collagen and elastic fibers (Fig. 1G).
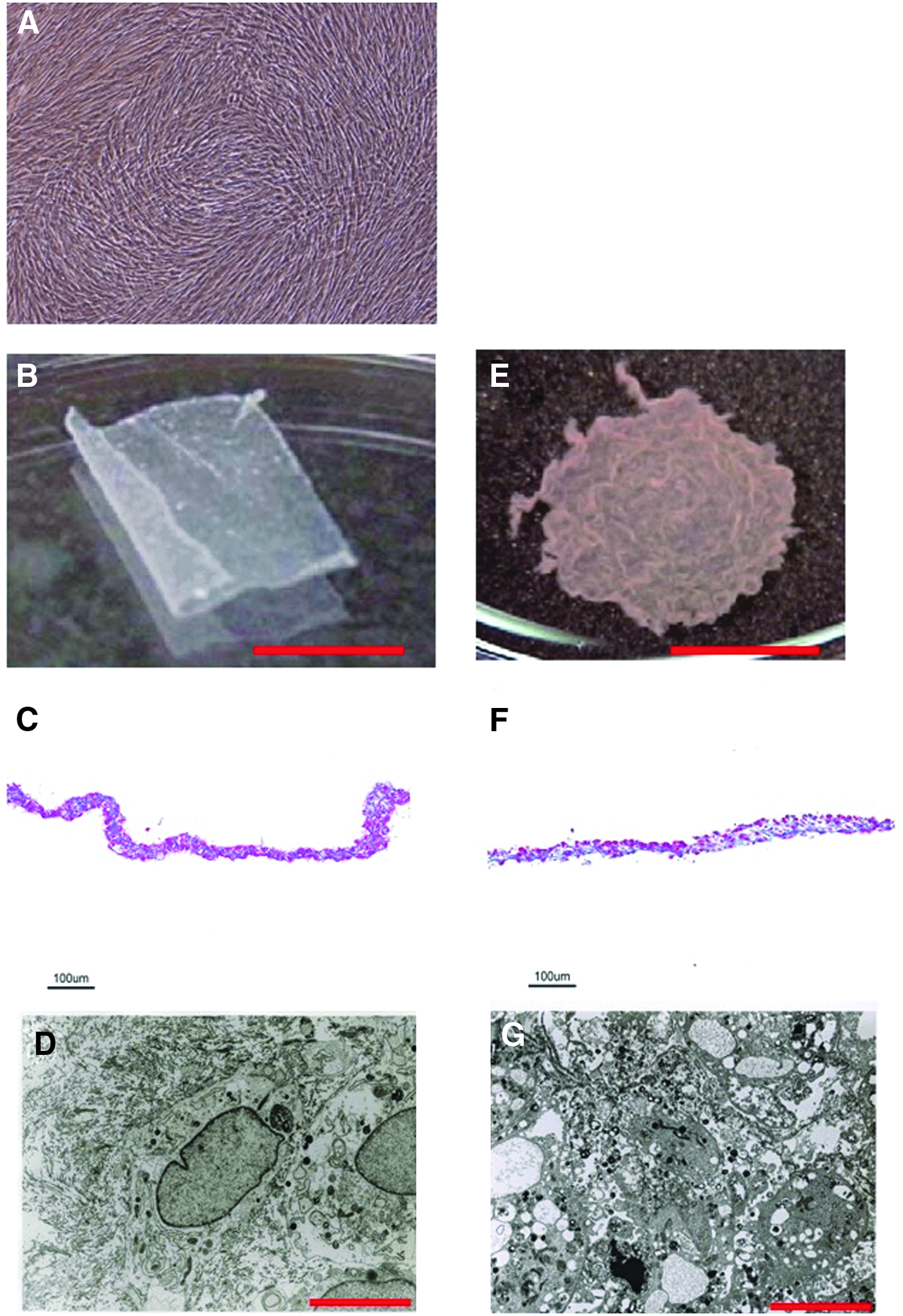
Fabrication of off-the-shelf FSs.
Secretion of growth factors from frozen–thawed FSs
The amounts of bFGF and VEGF released from frozen–thawed FSs were measured. bFGF levels significantly increased in frozen–thawed FSs compared with fresh FSs (490.8 ± 56.3 pg vs. 16.2 ± 13.2 pg, p < 0.01) (Fig. 2A). In contrast, fresh FSs increased secretions of both VEGF (137.3 ± 75.1 pg vs. 3196.7 ± 124.6 pg, p < 0.01) (Fig. 2B) and HGF (not detected vs. 448.3 ± 203.4 pg) compared with frozen–thawed FSs (Fig. 2C).

Secretion of growth factors from frozen–thawed FSs. Levels of cytokine secretion from frozen–thawed FSs and control fresh FSs were measured by enzyme-linked immunosorbent assay.
Cytotoxicity of the frozen–thawed process on FSs
The amount of LDH released from frozen–thawed FSs was significantly higher than that from fresh FSs (40.6 ± 23.8 U/L vs. 5.2 ± 2.9 U/L p < 0.05) (Fig. 3). After reculture, growth of fibroblasts in frozen–thawed FSs was observed to be the same as that in fresh FSs (Fig. 4).
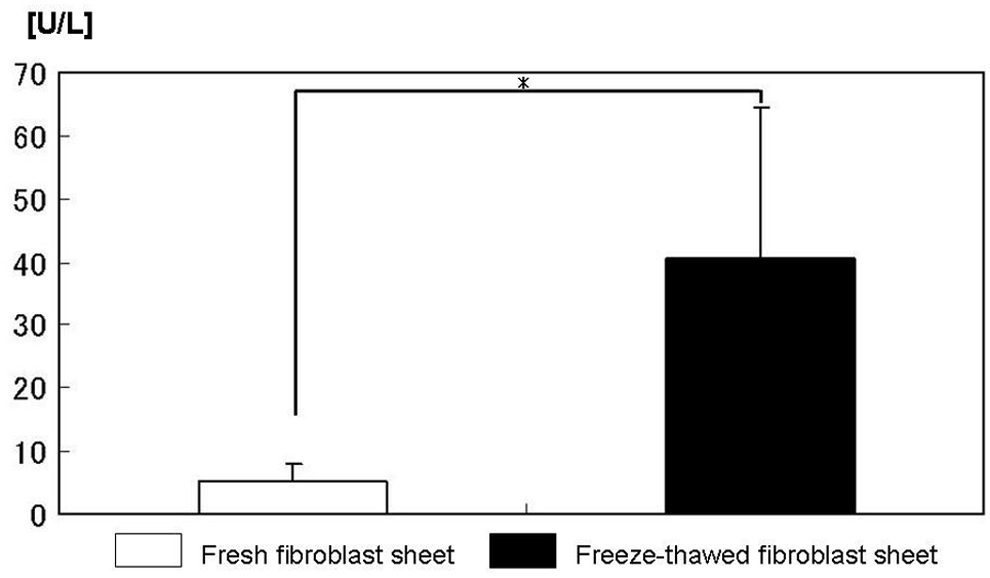
Cytotoxicity of a frozen–thawed FS. The amount of LDH released from the frozen–thawed FS increased significantly compared with the control fresh FS (40.6 ± 23.8 U/L vs. 5.2 ± 2.9 U/L). The bars indicate SD (p < 0.05). LDH, lactate dehydrogenase.
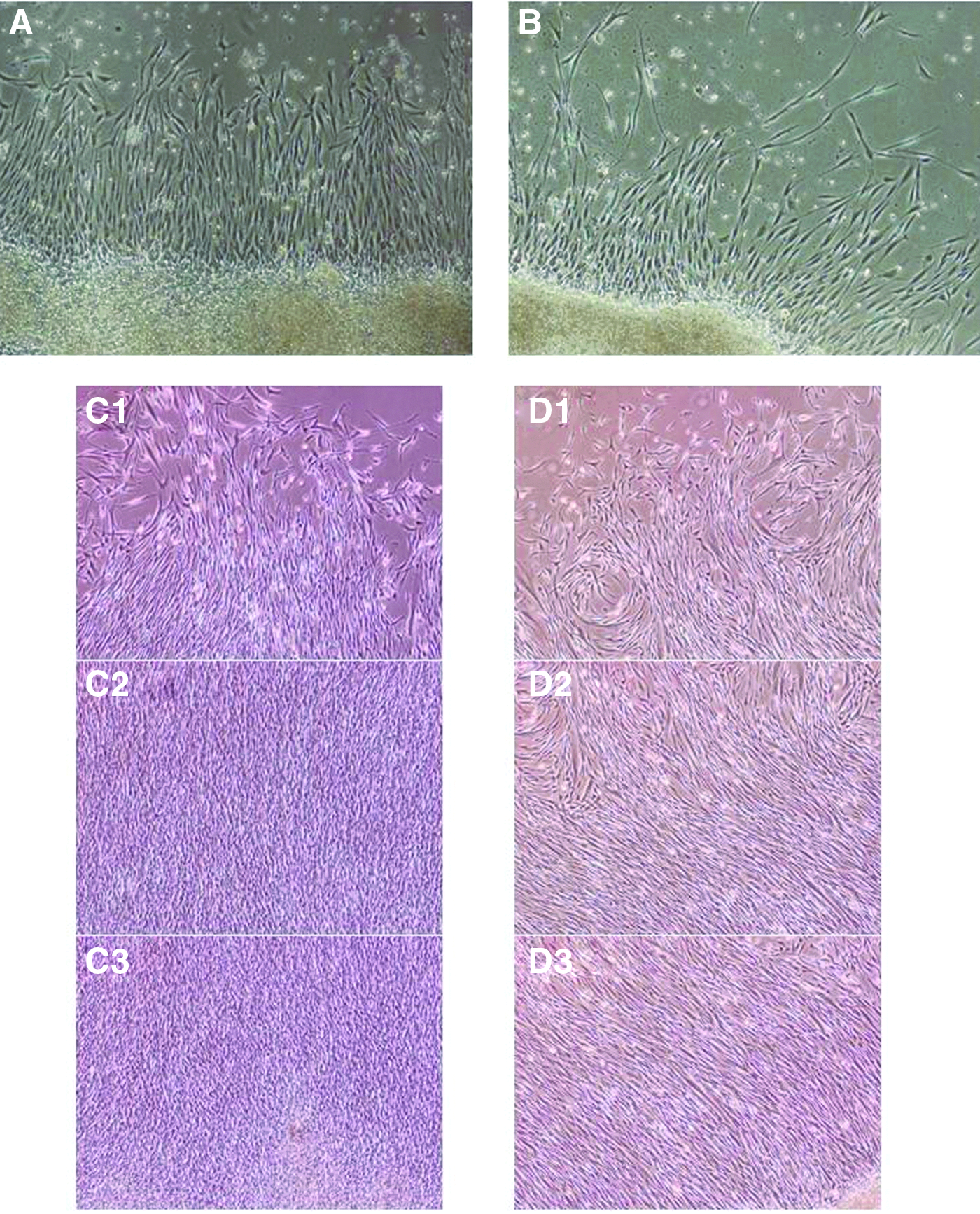
Growth of fibroblasts in frozen–thawed FSs. Phase-contrast micrographs of recultured frozen–thawed FSs at various stages. After reculture, growth of fibroblasts in the frozen–thawed FS resembled that of the control fresh FS.
Transplantation of off-the-shelf FSs for closing visceral pleural injuries
After creating a pleural injury with an intraoperative LAL, the frozen–thawed FS was then transplanted directly to the visceral pleural surface (Fig. 5A, B). Within 5 minutes, the frozen–thawed FS was confirmed to be attached stably over the pleural defect like a fresh FS without the use of sutures or fibrin glue (Fig. 5C). The transplanted frozen–thawed FS adhered to the pulmonary parenchyma surrounding the defect and the adhesive status was confirmed by the absence of air bubbles from the defect (Fig. 5D). Immediately after surgery, all LALs were completely sealed by frozen–thawed FSs.
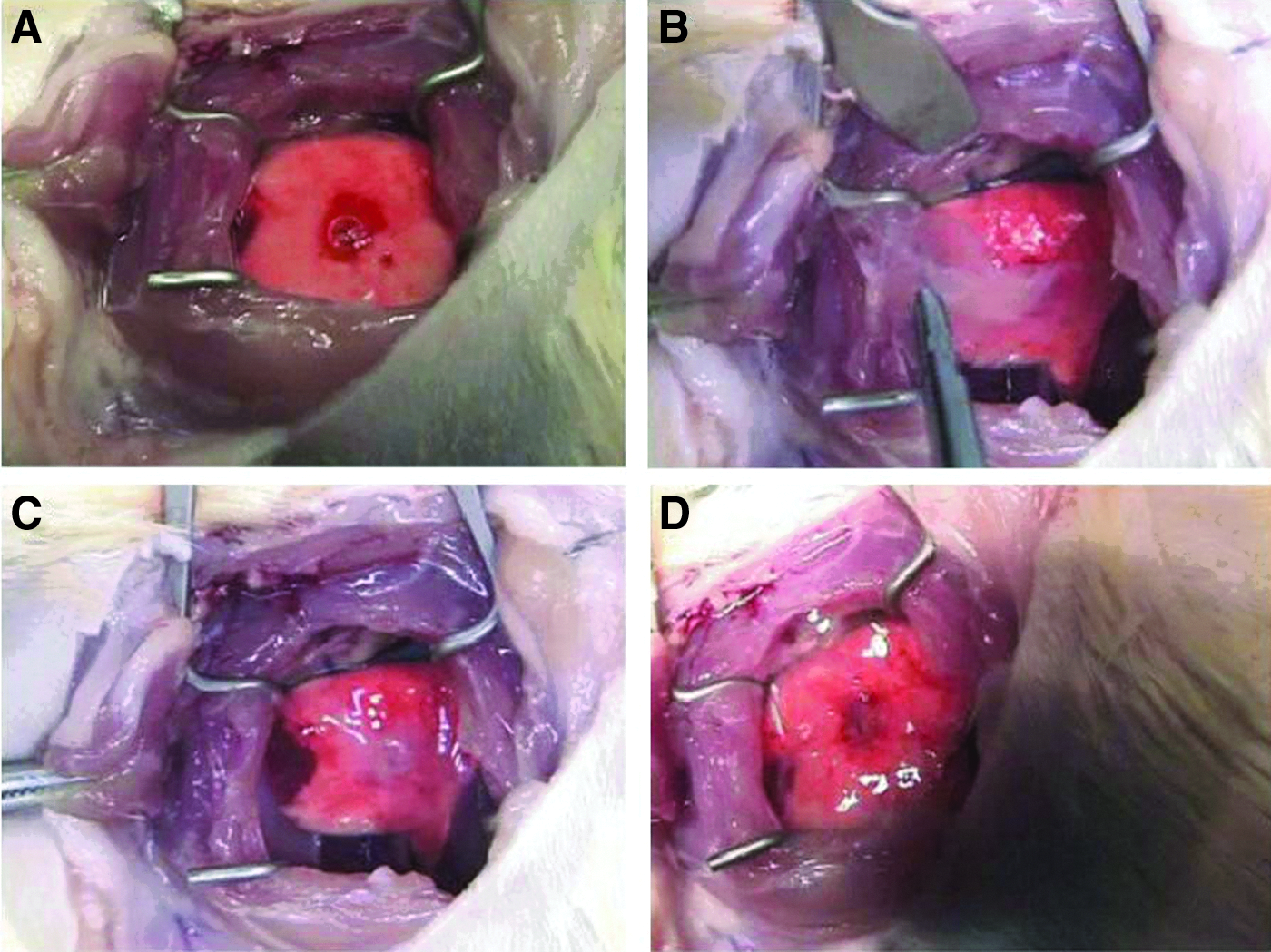
Transplantation of frozen–thawed FSs for closing visceral pleural injuries.
Frozen–thawed FSs provide a long-term and permanent seal for pleural defects
One month after the transplantation, frozen–thawed FSs maintained the closure of pleural defects without lung constriction in all rats and were flexible and elastic enough to follow lung expansion and contraction movements even under mechanical ventilation (Fig. 6). LALs were never observed. Histological examination revealed that the transplanted frozen–thawed FS was tightly attached to the pulmonary parenchyma with pleural defect and was significantly thicker with abundantly produced ECM and vacuolation, obviously the transplanted cell sheet had no signs of rejection, and no air space was found between the cell sheet and host lung tissues (Fig. 7).

Permanent sealing of a lung air leakage with a frozen–thawed FS. One month after transplantation of the frozen–thawed FS to seal visceral pleural defects, no air leakage was observed. The frozen–thawed FS was confirmed to close the pleural defect without any lung constriction. The white box indicates the location of the sealed pleural injury.
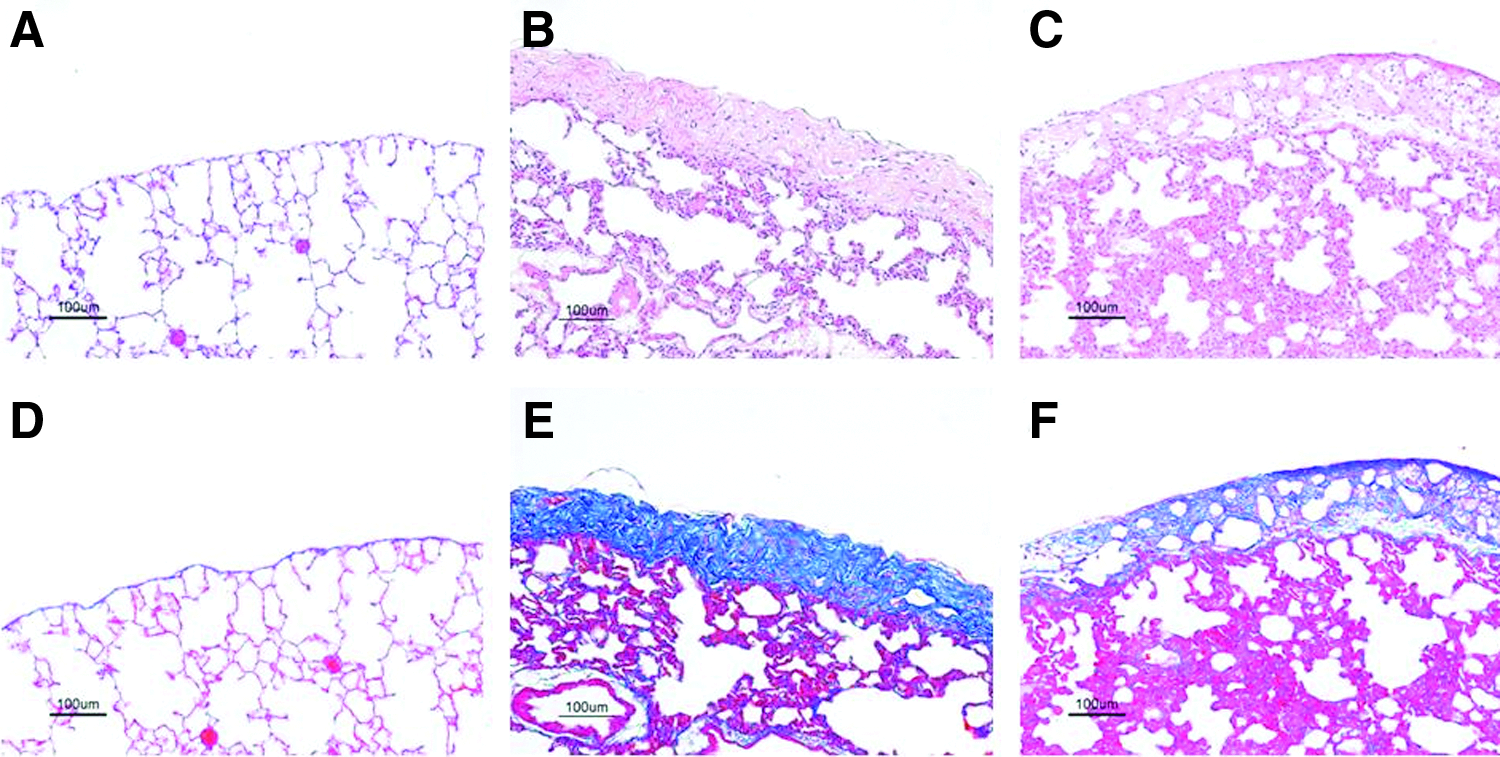
Histological analysis of frozen–thawed FSs 1 month after transplantation.
Discussion
LAL is the most common postoperative condition in patients who have undergone pulmonary resections.1–3,5 Postoperative LAL is caused by disrupted alveoli that are unable to be closed during the operation or have unpredictably reopened during the postoperative period. The wet surfaces of the postoperative lung spontaneously touch the parietal pleura for sealing the LAL. Conditions that interfere with pulmonary compliance, inflammation, and wound healing can prolong LAL. Therefore, intraoperative LAL prevention is the best approach. Many techniques previously advocated as methods for sealing LALs can only promote healing and prevent fistulization. Although various biological glues have been developed for intraoperative use, these many glues are absorbed easily, resulting in providing a weak ability to bond.15–19 These sealants were unable to function on the pleura because the pleura is a serous membrane lined with mesothelial cells, which form a monolayer of coffee bean-shaped cells having numerous microvilli on their surfaces, and eventually the microvilli cover the pleural surfaces. 14 The connective tissue layer in the visceral pleura has two important functions: (1) contributing to the elastic recoil of the lung, which is important for expelling air from the lung, and (2) restricting the volume to which the lung can be inflated. Mesothelial cells are recognized as composing a dynamic cellular membrane with many important functions, such as (1) the transport and movement of fluid and particulate matter across pleural surfaces, (2) leukocyte migration in response to inflammatory mediators, and (3) synthesis of cytokines, growth factors, and ECMs. Mesothelial regeneration involves migration of cells from the wound edge and attachment and incorporation of free-floating mesothelial cells from the pleural fluid onto the denuded pleural surface.20,21 The authors previously reported a novel and effective LAL sealant with tissue-engineered FSs,9–13 and FSs were confirmed to be durable, long-term LAL sealants that have sufficient flexibility for responding to dynamic movements of lungs during respiration and excellent biocompatibility by depositing ECM-enriched tissue on the lung surface. Transplanted FSs secrete a large amount of nonrigid ECM enriched with both type I and type III collagens. Deposition of an abundant and diverse ECM gives the cell sheet sealants sufficient flexibility and elasticity that are required to expand and contract for responding to the lung movement during normal respiration, and at the same time, the ECM can also maintain stable closure of air leaks. Creation of human FSs requires a 3-week cultivation period before surgery, and fresh FS clinical applications such as for emergency surgery and unexpected surgery are limited by problems relating to mass production, preservation, and transportation. For solving these problems, therefore, frozen–thawed FSs are more practical for clinical use such as for treating intraoperative LALs.
Alternative preservation methods such as cryopreservation may prove to be acceptable. 22 Cryopreservation is one of the methods used for preserving grafts and is now widely used as a long-term preservation technique for biological tissues, and the method is believed to be the best method for maintaining graft viability. 23 The present study investigated possible cellular damages, which were due to cryopreservation and would significantly affect its clinical applications. Although fresh FSs released VEGF, bFGF, and HGF and off-the-shelf FSs could also release bFGF and VEGF, the causes of remarkable decreases in VEGF and nondetectable HGF were unclear. Sawa and Kuroyanagi have reported that (1) an allogeneic cultured dermal substitute (CDS) is then cryopreserved from 1 to 6 months, (2) amounts of VEGF and HGF are measured, and (3) amounts of these growth factors released from recultivation after thawing decrease more than those before cryopreservation. Their study concluded that EGF precipitates on the CDS upon release of VEGF and HGF, and the results are similar to this study's results.14,24,25 However, an increase in bFGF production in off-the-shelf FSs was found to be significantly higher compared with fresh FSs. Strandberg et al. found that the freeze–thaw procedure results in ∼4- to 10-fold enrichment of bFGF, and their experimental results supported the results of this study. 26 Furthermore, the amounts of LDH released from frozen–thawed FSs were higher compared with fresh FSs. These results suggested that the large amount of LDH released from frozen–thawed fibroblasts is a result of possible cellular damage through the freezing and thawing processes, although cryopreservation-induced necrosis was found in a small percentage of cells and frozen–thawed fibroblasts were found to be still alive after reculture. Since bFGF regulates early development and organogenesis, frozen–thawed FSs rich in bFGF may be highly beneficial for repairing pleural injuries.27–30 In addition, transplanted frozen–thawed FSs can not only attach stably and flexibly to the wet surfaces of a wounded lung for quickly sealing intraoperative LALs but can also keep their dynamic status responding to the movement of contraction and relaxation during respiration with permanent closing of the pleural defects. The sheets also integrate with native host tissue similar to fresh FSs. These results suggested that cells in off-the-shelf FSs were able to show sufficient abilities to proliferate and release various cytokines. Frozen–thawed FSs were effective for closing LALs during pulmonary surgery as an off-the-shelf technique and were able to be used as a pleural substitute.
Footnotes
Acknowledgments
This study was supported, in part, by the Grant-in-Aid for Scientific Research (C) (21591827) and Formation of Innovation Center for Fusion of Advanced Technologies in the Special Coordination Funds for Promoting Science and Technology from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan.
Author Disclosure Statement
Dr. M.Y. is a stakeholder of CellSeed, Tokyo, Japan. No competing financial interests exist for the remaining authors.