
Abstract
Context:
In the National Cerebral and Cardiovascular Center (NCVC) Biobank, buffy coats have been collected from patients and stored with cryoprotective agents as a possible source for viable blood cells, using cost-efficient methods for storage. However, whether viable cells for in vitro studies can be recovered from these biospecimens has not been verified.
Objective:
To investigate whether T cells can be collected and expanded as viable cells from cryopreserved human buffy coats.
Design:
After thawing of cryopreserved buffy coat specimens, CD3-positive cells were isolated from the cell suspension using a leukocyte separation filter coated with an anti-CD3 antibody, and the filter-attached cells were cultured in T cell culture medium. To analyze the characteristics of these cultured cells, histocytological analyses of Giemsa staining, immunocytochemical (ICC) staining for CD3, and flow cytometry for CD3 in live cells were conducted.
Results:
A few days after starting cell culture, cell clusters were observed, and they gradually grew in size. Using Giemsa staining, the expanded cells were found to be ∼15 μm in diameter, having round nuclei, a high nucleus/cytoplasm ratio, and cytoplasm stained light blue, which is characteristic of lymphocytes. From ICC staining, these cells were CD3 positive, a pan-T cell marker among lymphocytes. Furthermore, CD3 immunoreactivity in live cells was detected in a flow cytometry assay, though that for CD19 was not detected, which is a marker of pan-B cells.
Conclusions:
These results suggest that T cells can be expanded from buffy coats cryopreserved at −180°C as an adequate method of NCVC Biobank, highlighting these biospecimens as a possible useful source for future in vitro studies.
Introduction
The National Cerebral and Cardiovascular Center (NCVC) Biobank has collected and stored various human biospecimens, including organ tissue, serum, plasma, buffy coat, and buffy coat-derived DNA, at −80°C for in vitro medical studies. In addition, buffy coats have been collected and stored at −180°C in a liquid nitrogen storage system as a source of viable blood cells.
In some biobanks, peripheral blood mononuclear cells (PBMCs) have been collected and stored as viable blood cells.1–3 PBMCs are blood cells with a single round nucleus, and they include lymphocytes (T cells, B cells, and NK cells), monocytes, and dendritic cells. PBMCs represent an invaluable cell resource for various studies related to the immune system, including vaccine development, drug discovery, cancer treatments, and the establishment of cell lines.4,5 Recently, it has been reported that induced pluripotent stem (iPS) cells were generated from cryopreserved PBMCs.6–9 PBMCs are usually isolated from whole blood via density gradient centrifugation using a commercial gradient medium, such as Lymphoprep and Ficoll, or specialized tubes, such as the BD Vacutainer® CPT™. However, the methods for collecting PBMCs are expensive and complicated, making them unsuitable for biobanks, which collect multiple specimens daily.
Therefore, we focused on the buffy coat, which contains lymphocytes, monocytes, and granulocytes and can be collected simply and cost-efficiently without specialized reagents, such as a gradient medium. We considered the cryopreservation of buffy coats to be a useful and cost-effective approach for collecting viable blood cells. Then, to prevent crystal formation, which destroys cells during the freezing process, we cryopreserved the buffy coat using CP-1 cryopreservation medium. The CP-1 medium has been used for the cryopreservation of cells, such as peripheral blood and umbilical cord blood cells.10–12 CP-1 medium does not require rate-controlled freezing. However, its use for buffy coat cryopreservation has not been studied. Therefore, it is necessary to confirm whether buffy coat cryopreserved using CP-1 medium can be useful as a source of viable cells for in vitro studies. T cells are the major cell population of the immune system present in the buffy coat. And recently, it has been reported that T cells could be important cell sources for generating iPS cells, because peripheral blood including T cells can be collected with minimal invasion and easily proliferate in vitro using anti-CD3 antibody and interleukin-2 (IL-2).13–15 Therefore, in this study, we investigated whether T cells can be collected constantly and expanded as viable cells from cryopreserved buffy coats.
Materials and Methods
Collection of samples
Peripheral blood samples were obtained from 13 patients who consented to the submission of their samples to the NCVC Biobank after reading a handbook and leaflet explaining the purpose. The study was approved by the Institutional Review Board of the National Cerebral and Cardiovascular Center (Approval No. M23-068-9). The blood samples (1.8 mL blood into tubes containing 0.2 mL sodium citrate buffer) were obtained from patients with cerebral and cardiovascular diseases, but without active malignant diseases. After the plasma of whole blood was used for a blood test, the residual blood was kept at room temperature and used as follows within 24 hours. About 0.5 mL of the blood cell layer containing the buffy coat and 0.5 mL of the plasma were gently mixed with 0.5 mL of CP-1 cryopreservation solution (Kyokuto Pharmaceutical Industrial Co., Ltd, Tokyo, Japan) consisting of 12% hydroxyethyl starch and 10% dimethyl sulfoxide (DMSO). Then, 1.5 mL of the mixture was aliquoted into two 2-mL cryovials (750 μL) each and immediately placed in a −80°C freezer. After freezing, the vials were stored in a −180°C liquid nitrogen tank until use.
Isolation and culture of T cells
The cryovials stored in liquid nitrogen were removed, and quickly warmed in a water bath at 37°C for ∼1 minute until the wall of the sample melted, resuspended in 10 mL of prewarmed cell medium (Dulbecco's modified Eagle's medium [DMEM] containing 10% fetal bovine serum [FBS], 15 U/mL heparin, and 100 U/mL DNase I) at 37°C, transferred to 6 cm dishes, and incubated in a 5% CO2 atmosphere at 37°C for 1–2 hours to allow for cell recovery. After the incubation period, 4–5 mL of the supernatant (containing mostly cell debris and ghost red blood cells) was removed, and 5–6 mL of the remaining solution (containing mostly cells) was passed through a LeukoCatch II filter (WATSON Co., Ltd., Kobe, Japan) coated with 10 μg/mL of anti-CD3 antibody (clone: OKT3; eBioscience, San Diego, CA). The filter with attached cells was transferred to a 6-well culture plate and cultured in culture medium (AIM V medium [Gibco Gaithersburg, MD] containing 2.5% FBS and 300 U/mL IL-2) in a 5% CO2 atmosphere at 37°C. Every 2 or 3 days, 1 mL of culture supernatant was replaced with 1 mL of fresh culture medium (the final IL-2 concentration was 300 U/mL). As the cells proliferated, they began to spontaneously detach from the filter. After gently pipetting the filter to allow the cells to detach more easily, the filter was temporarily removed, and photomicrographs of cells were taken. When the cells had proliferated sufficiently, the cell suspension was centrifuged for 10 minutes at 1000 rpm at 4°C, the supernatant containing ghost red blood cells and cellular debris was carefully removed, and the pellet was gently dissociated using a pipette, transferred into new wells coated with 10 μg/mL anti-CD3 antibody according to the degree of cell proliferation, and cultured in a 5% CO2 atmosphere at 37°C.
Cell viability
After the cells were resuspended in cell melting medium as described previously, they were incubated in phosphate buffered saline solution (PBS) containing 4.5 μM propidium iodide (PI; Dojindo Laboratories, Kumamoto, Japan), 2.0 μM calcein-AM (Dojindo), and 10 μM Hoechst 33342 (Dojindo) for 60 minutes at 37°C. Calcein-AM (green), PI (red), and Hoechst 33342 (blue) were used to stain the cytoplasm of viable cells, the nuclei of dead cells, and the nuclei of all cells, respectively. A drop of the suspension was placed on a hemocytometer slide, and fluorescent images were observed and taken using a BZ-X800 fluorescence microscope (Keyence, Osaka, Japan). Cell viability was calculated as a percentage of calcein-AM-stained cells among all Hoechst 33342-stained cells.
Giemsa stain for cell morphology
Cells were washed with PBS and fixed immediately in CytoRich Blue (Becton, Dickinson and Company [BD], Franklin Lakes, NJ). These cells were transferred onto a glass slide via the cytocentrifugation method (cytospin, 800 rpm, 2 minutes). After air-drying, the slides were stained with Giemsa stain. In brief, the slides were immediately stained with May–Grünwald stain solution (Muto Pure Chemicals Co., Ltd., Tokyo, Japan) for 5 minutes. Next, the slides were washed in tap water and immediately stained with Giemsa stain solution for 20 minutes. Next, the slides were washed in tap water and immediately allowed to air dry for 20 minutes. The slides were preserved using glass coverslips and mounting medium (Merck KGaA, Darmstadt, Germany).
Immunocytochemistry
After the specimens were processed as described previously, the slides were fixed with Merckofix spray (Merck). CD3 staining was performed on an automated Leica Bond III (Leica Microsystems, Melbourne, Australia) according to the manufacturer's instructions. Antibody detection and counterstaining with hematoxylin were performed using a Bond Polymer Refine Detection Kit (DS9800; Leica Biosystems). In brief, the slides were incubated with a primary antibody against CD3 (Dako, Glostrup, Denmark), followed by incubation with a secondary antibody, and then the slides were incubated with goat-horseradish peroxidase polymer and 3,3′-diaminobenzidine substrate. Images were taken using a microscope (ECLIPSE Ni-U; NIKON, Japan).
Flow cytometry
Cells were harvested, and the cell suspension was incubated in spillover Blocking Reagent (MACS Miltenyi Biotec, GmbH) to inhibit nonspecific or Fc receptor-mediated binding of CD3 antibody. Each cell sample was stained with fluorescein isothiocyanate (FITC)-conjugated anti-CD3 antibody (BD), FITC-conjugated anti-CD19 antibody (eBioscience), and a live/dead dye (7-amino actinomycin D [7-AAD]; BD), and the expression of each marker was measured using FACSVerse (BD). FITC-conjugated mouse IgG2a kappa antibody (BD) and FITC-conjugated mouse IgG1 kappa antibody (eBioscience) were used as the isotype control.
Results
The median viability of cells isolated from the buffy coat was 76%, and the median number of viable cells was 3.5 × 106 cells, indicating that blood cells with nuclei were present in cryopreserved buffy coat, allowing their potential use and expansion.
Next, to verify whether these viable blood cells could be expanded, the ability of expand T cells from buffy coat was examined. T cells, which represent the major cell population among blood cells, express CD3 antigen. Approximately 5 days after starting the cell culture, some clusters of cells formed (Fig. 1A). At 13 days, these clusters were widely observed (Fig. 1B) and gradually grew larger (Fig. 1C). After 16 days of culture, these cell clusters were disrupted using a pipette, transferred into new wells coated with anti-CD3 antibody, and further cultured (Fig. 1D). These cells continued to expand while forming cell clusters (Fig. 1E), suggesting that these cells were activated lymphocytes. 16 The number of cells after 23 days of culture was ∼1 × 107. However, the cells that did not form clusters proliferated from ∼25 days (Fig. 1F).
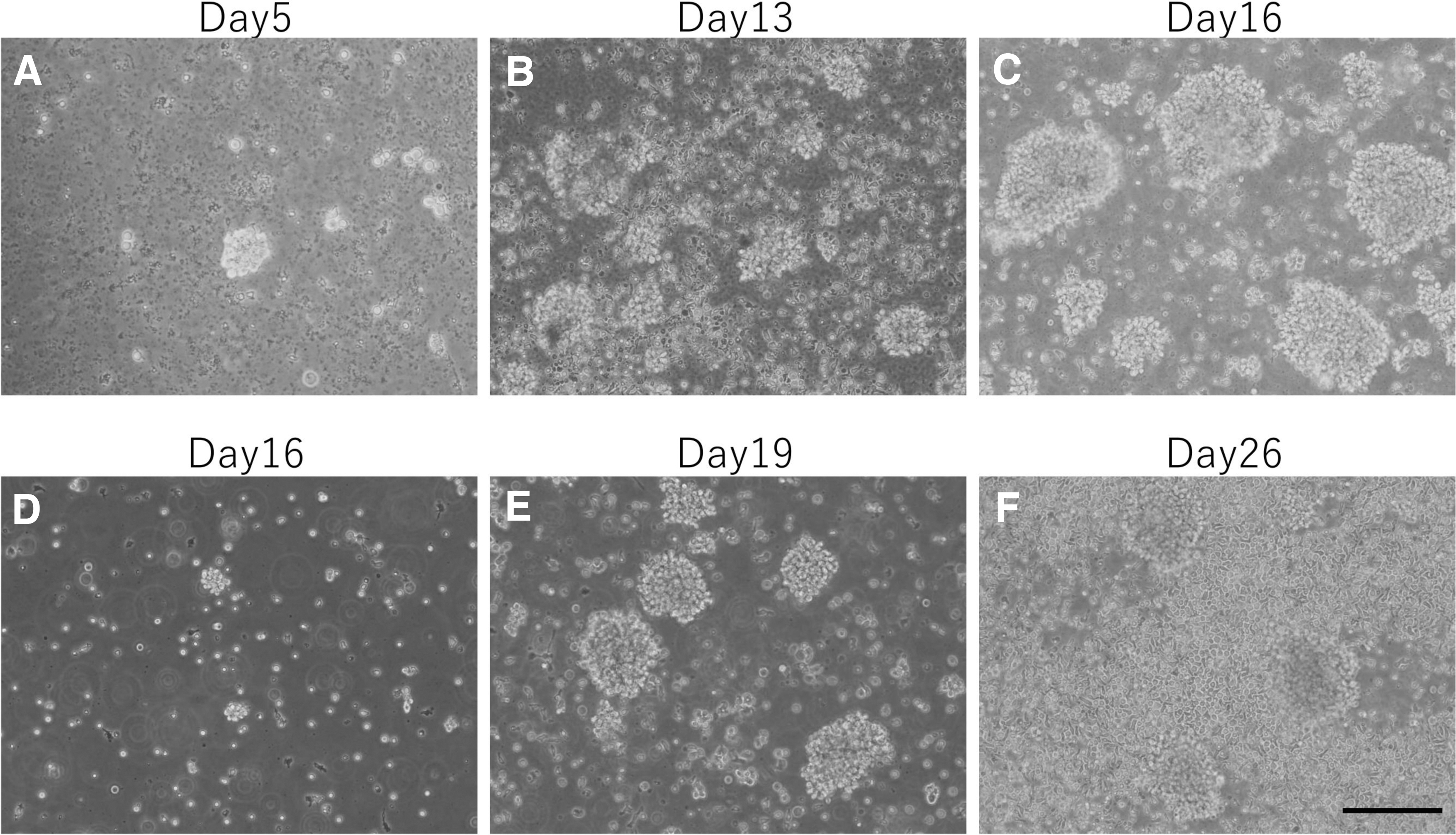
Phase-contrast photomicrographs of cells cultured for 5
To identify the characteristics of these expanded cells (including both clustering and nonclustering cells), histological analysis using Giemsa staining was conducted after 23 days of culture. These expanded cells were ∼15 μm in diameter. The nuclei were round, the nucleus/cytoplasm ratio was high, and cytoplasm stained light blue (Fig. 2A). These results suggested that these cells were lymphocytes, but this analysis could not distinguish the types of lymphocytes (B cells, T cells, or NK cells). Therefore, to further evaluate the phenotypic properties of these cells, immunocytochemical staining for CD3 was performed after 23 days of culture. Because CD3 is an antigen that is expressed at all stages of T cell development and it is expressed by both resting and activated T cells, it has been used as a pan-specific marker for T cells. 17 As a result, it was observed that cells of >90% were positive for CD3 (Fig. 2B). To further confirm the expression of CD3 in these cells, flow cytometry for CD3 was performed, the proportion of live cells (7-AAD-negative cells) is 99.44% (Fig. 2C) and the proportion of CD3-positive cells was 95.61% (Fig. 2D). Furthermore, flow cytometry for CD19 (B cell marker) was performed, but the proportion of CD19-positive cells was 0% (Fig. 2E).
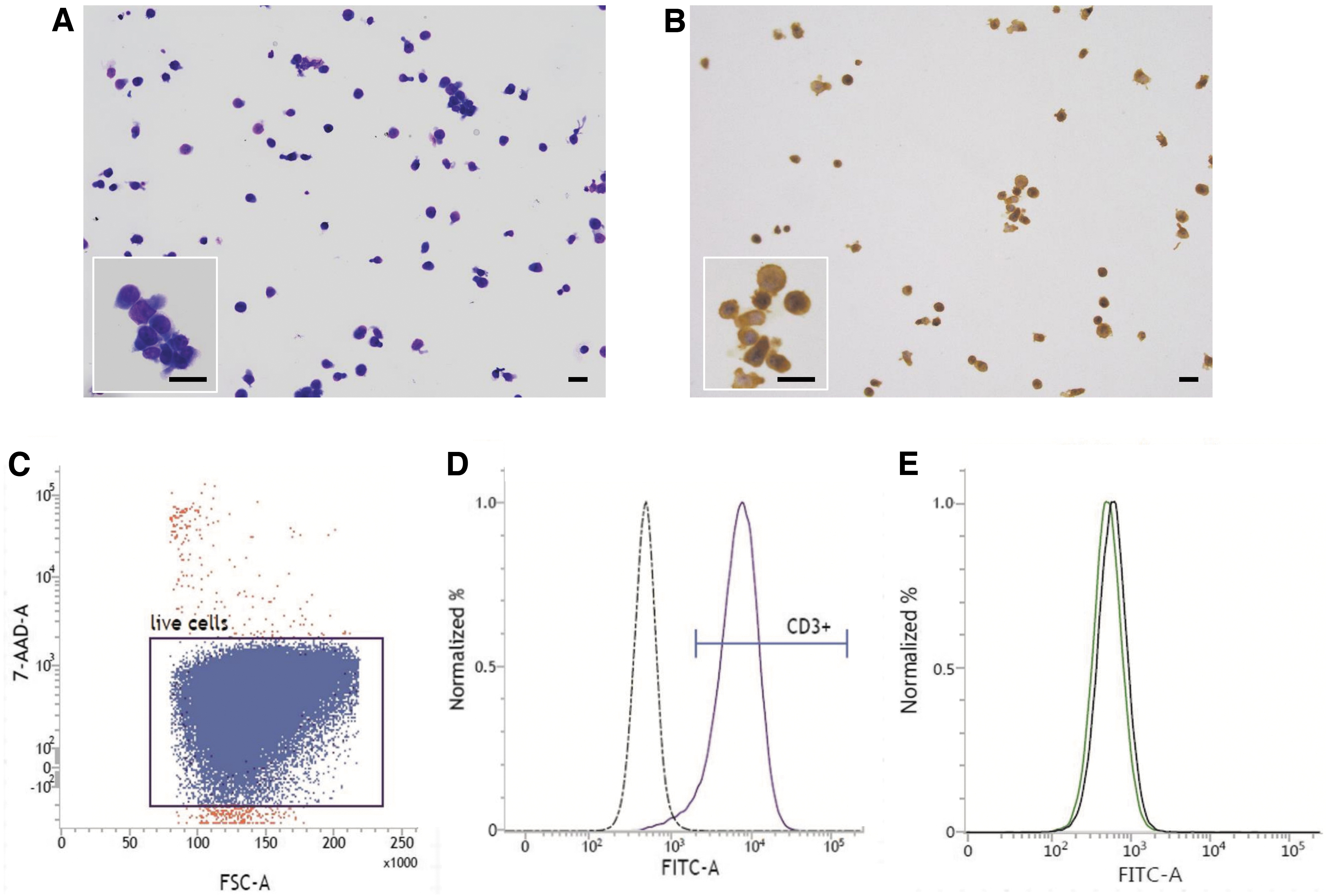
Characterization of expanded cells. Giemsa staining
Discussion
Our data indicate that T cells can be collected and expanded as viable cells from cryopreserved buffy coat in the NCVC Biobank. The NCVC Biobank has routinely collected and stored buffy coats from >2000 patients a year who suffer from cerebral and cardiovascular diseases. And it has been intended that the cryopreserved buffy coats will be used as viable cells for in vitro studies as needed. PBMCs are usually isolated from blood by centrifugation using specialized tubes that include density gradient media. After the centrifugation, the PBMC layers are harvested, washed with PBS, centrifuged again to remove residual media, and the PBMCs are harvested and stored with cryoprotective agents in freezer. By contrast, in the NCVC Biobank, using general blood collection tubes, the buffy coat is harvested from citrate blood collection tubes by centrifugation and promptly stored in a freezer with the addition of cryoprotective agents. This method is an advantage in cost and efficiency, especially in biobanking of blood samples from patients with rare cardiovascular diseases, among daily outpatient settings and intensive care wards, considering only a small fraction of biobank samples that would be used for further in vitro assays. Although such cryopreserved buffy coats must be thawed, recovered, and expanded for subsequent assays, these storage methods could be suitable for the collection of blood samples from patients with rare cardiovascular diseases, especially in daily outpatient settings, as well as in intensive care wards.
To our knowledge, little research has examined the cryopreserved buffy coat as a source of viable blood cells for in vitro medical research studies. A report by Meraviglia et al. indicated that cryopreserved buffy coat can be used to culture PBMCs and generate iPS cells from PBMCs. 18 However, in their study, they used freezing medium containing 90% FBS and 20% DMSO, and they did not elucidate which PBMC subtype was cultured.
In this study, some points must be elucidated regarding the use of cryopreserved buffy coat. (1) When the content of the cryovials was first thawed in a water bath at 37°C and resuspended in the resuspension medium (DMEM containing 10% FBS), the formation of a fibrin-like gel was observed. To prevent this, 15 U/mL heparin was added to the resuspension medium. In addition, 100 U/mL DNase I was added to the resuspension medium to degrade extracellular DNA from broken cells. (2) A natural standing separation process was adopted instead of centrifugation, to prevent the formation of solid cell pellets because centrifugation can result in cell damage. (3) Furthermore, we used LeukoCatch II filters to separate leukocytes from whole blood cells without centrifugation. 19 The cells proliferated better onto the anti-CD3 antibody-coated filters than the noncoated filters (Supplementary Fig. S1). This result indicated that coating the filters with anti-CD3 antibody provides a proliferation-stimulating surface and may also be effective for T cell isolation. However, in the early stages of cell culture, we were unable to develop a method to detach the adherent cells from the filters. Therefore, it was not possible to determine the extent to which the anti-CD3 antibody contributed to the efficient recovery of T cells. Centrifugation was adopted when a sufficient number of cells were obtained. Then, it became possible to remove ghost red blood cells, especially the cellular debris.
In this study, the T cell isolation method was simple and cost-effective but time-consuming. Conversely, it has been reported that although isolation methods using antibody-coated magnetic beads are easy and speedy and allow highly specific isolation, they are expensive.20,21 Therefore, further experiments are required to examine whether these methods can be used to separate T cells from cryopreserved buffy coat.
Various culture media that have been developed for T cells have different effects on the expansion and phenotypes of the cells.22–24 In this study, we selected AIM V medium containing IL-2 and serum. Originally, although AIM V medium was designed for use as a serum-free medium, the cell number was not sufficient in the absence of serum, in line with a previous report. 22 Using this culture medium, activated cluster-forming cells were observed after ∼7 days of culture; however, nonactivated cells that did not form clusters proliferated after ∼1 month. Therefore, to ensure the long-term maintenance of activated T cells, anti-CD3/CD28 beads might be necessary to efficiently activate and expand T cells.25,26 These results suggest a need for each researcher to choose the most appropriate culture method based on the study goals.
The temperature of cryopreservation may affect cell viability and recovery.27,28 In our study, the viability of cells after cryopreservation at −80°C for ∼1 year was almost similar (65%) and did not appear to affect the expansion (Supplementary Fig. S2). Furthermore, it is known that cryopreservation and thawing methods can affect cytokine production, the expression of cell surface molecules, and the viability of PBMCs and T cells,29–36 but elucidation of their effects will require additional research.
The study findings suggest that buffy coat cryopreserved in the NCVC Biobank can be collected and expanded as living T cells, which could be a source for future in vitro research. Applicability of this method to blood samples from patients with a wide variety of diseases, including malignancies, needs further investigation.
Footnotes
Acknowledgments
We thank all members of the NCVC Biobank, Dr. T. Ishibashi, and Mrs. S. Arata for their excellent technical support and also thank Mrs. K. Yamazaki for administrative support.
Author Disclosure Statement
No conflicting financial interests exist.
Funding Information
This research was supported by the “Platform Program for Promotion of Genome Medicine” of Japan Agency for Medical Research and Development (AMED).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.