
Abstract
Background:
Decellularized human nerves overcome the limitations of the current treatments for large peripheral nerve injuries. However, the use of decellularized nerves requires an “off-the-shelf” availability for useful and actual clinical application. In this study, we addressed the preservation of the native and decellularized human nerve matrix in an integrative approach for tissue scaffold production.
Materials and Methods:
For native nerve matrix preservation analysis, we used histological examination and immunofluorescence to examine the structure, biomechanical assays to evaluate the tensile strength and Young's modulus, and analyzed the extracellular matrix (ECM) composition using enzyme-linked immunosorbent assay (ELISA) and biochemical assays for laminin, collagen and sulfated glycosaminoglycans (sGAG). After decellularization, nuclear remnants and DNA content were evaluated using 4′,6-diamidino-2-phenylindole (DAPI) staining and the picogreen quantification assay, as well as immunofluorescence or ELISA for cell rests (S100 protein and myelin staining) evaluation. Decellularized cryopreserved scaffolds were assayed for biomechanics, ECM composition, and structural maintenance. Cytotoxicity assays were performed to evaluate the biocompatibility of the nerve matrix extracts after cryopreservation.
Results:
We compared different strategies for native nerve storage and found that preservation up to 7 days at 4°C in Roswell Park Memorial Institute medium maintained biomechanical properties, such as Young's modulus and tensile strength, along with the structure and ECM composition, regarding laminin, collagen, and sGAG. After a successful decellularization process, that eliminated cell remnants, nerve scaffolds were frozen in an “in house” formulated cryoprotectant, using an automatic controlled rate freezer. Nerve structure, ECM composition, and biomechanical properties were maintained before and after the freezing process in comparison with native nerves. The extracts of the nerve scaffolds after thawing were not cytotoxic and the freezing process sustained good viability in 3T3 cells (graphical abstract).
Conclusion:
Since our approach facilitates transport, storage, and provide a ready-to-use alternative, it could be used in a clinical application for the treatment of long-gap peripheral nerve injuries in regenerative medicine.
Introduction
Tissue and organ decellularization have attracted attention in the field of regenerative medicine because of the lack of enough donors to cover the current needs. 1 Decellularization involves several processes and strategies to obtain immunogenic free scaffolds from native organs or tissues, while preserving the extracellular matrix (ECM) composition and biomechanical properties. 2 Therefore, decellularized tissues and organs can decrease the number of patients needing a transplantation because they lessen organ specificity. In fact, decellularization is commonly used in clinical applications of reconstructive surgery such as transplantation of decellularized heart valves, 3 treatment of large burns with decellularized dermal matrix, 4 and soft tissue filling or other applications. 1 Specifically, decellularization of human peripheral nerves is innovative and its research application responds to the limitations and drawbacks of the current treatments for long-gap peripheral nerve injuries.
When there is a loss of tissue in a peripheral nerve, the organs and tissues that are innervated by this nerve lose their functions. If left without treatment, this injury causes a permanent disability and it is estimated that these kind of lesions affect 1 per 1000 habitants in Europe, most prominently adults incorporated in the employment system. 5 The current gold standard treatment is autotransplantation, where commonly a sensitive nerve is transplanted to regenerate a motor nerve. However, the outcomes are limited because sensory nerves express specific regenerative signals for fibers of sensory nerves and not for the fibers of motor nerves, 6 so the risk/benefit ratio of the surgical procedure can be compromised. Besides, the autografts that can be extracted do not always match with the damaged nerve in terms of size and caliber, and this limits the donor tissue available. 7 In contrast, allografts can be available in the size or caliber required, although allotransplantation offers lower successful clinical results 8 and requires the use of immunosuppressants to avoid immune rejection. 9
Therefore, there is a need to produce decellularized nerve scaffolds for nerve regeneration. This process implies various phases. The first step is the preservation of the native tissue until it can be processed, which is important to schedule with enough prevision in tissue banking. This is followed by the decellularization procedure itself and is continued with a final step that comprises the preservation of the decellularized matrix for long-term storage. In these steps, the maintenance of the structure and composition of the basement membrane is essential. This is formed by endoneurial tubes that are mainly composed of laminin and collagen, through which the nerve fibers grow during the regeneration process. 10 Disruption or damage in the endoneurial tubes would hinder the growth of the nerve fibers and would, therefore, inhibit the nerve regeneration and functional restoration.11,12 Furthermore, the alteration of the structure of the endoneurial tubes can lead to pathological formation of neuromas. 13 In addition, the maintenance of the biomechanical characteristics is essential for proper functionality restoration and also for scaffold handling during surgery. 14 Importantly, preservation at low temperatures could lead to the formation of ice crystals within the tissue, jeopardizing the biomechanical and structural properties. Thus, not only the decellularization methodology, but also the methods used to preserve the nerve scaffold should not change the properties of the matrix, to ensure clinical outcomes.
Finally, the decellularized nerve matrix should be an “off-the-shelf” product, easier to store, and cheaper to ship for clinical needs. The overall process cannot be excessively expensive to support, so its cost-effectiveness can be maintained under a threshold, making the production affordable within the health care system. Hence, it is of utmost importance to study the preservation steps pre- and postdecellularization.
In this study, we study different strategies to preserve human native nerves until tissue processing, as well as the cryopreservation after the decellularization process. To do so, we analyzed properties such as structure, composition, and biomechanical properties of the nerve matrix at these steps, and compared them with the properties of the native nerve.
Materials and Methods
Ethical considerations
This research followed the ethical precepts of the Declaration of Helsinki (Fortaleza, Brazil, October 2013) and was accepted by our local ethics committee (CEIC, Hospital Clinic de Barcelona; Ref 7365-12). Human samples were obtained, processed, and analyzed according to current guidance in relation to the collection and preservation of human tissues for clinical use (EEC regulations 2004/23/CE and 2006/17/CE) and in accordance with the protocol and legal requirements for the use of biological samples and biomedical research in Spain (Law 14/2007 and RD 1716/2011). In addition, the acquisition, processing, and preservation of the tissues for this study are in accordance with the Spanish Laws for the Development and Applications of Organ Transplants (RD 9/2014). All the information provided before donation, together with informed consent, stressed that the samples obtained were to be used for clinical application and/or applied research. The use, protection, communication, and transfer of personal data complied with local regulations (Law 05/2018).
Peripheral nerves
Human sural nerves from eight deceased donors were obtained from the Barcelona Tissue Bank (BTB-BST, Barcelona, Spain; www.bancsang.net/en_index/) within 24 hours postmortem. The mean age of the donors was 45.5 ± 7.89 years and five donors were male, whereas three donors were female. Five donors died by brain death and three donors died by cardiac arrest.
Preservation predecellularization
Nerves were obtained from deceased donors within 24 hours. Native nerves were preserved as follows: (1) up to 7 days in Roswell Park Memorial Institute (RPMI) media without phenol (Gibco, Carlsbad, CA, USA) plus 1% antibiotics (vancomycin [Pfizer, MA, Spain], penicillin [Normon, MA, Spain], and streptomycin [Reig Jofre, BCN, Spain]) at 4°C; (2) frozen in the aforementioned media with dimethyl sulfoxide (DMSO) (WAK-Chemi medical GmbH, Steinbach, Germany) and human serum albumin (HSA; Albutein, Grifols, BCN, Spain), ratio 1:0.4; (3) frozen in the aforementioned media with trehalose (Life Sciences Advanced Technology, St. Petersburg, FL, USA) and HSA (Grifols), ratio 1:0.4. The samples were frozen in an automatic controlled rate freezer (Cryoson 2010; Cryoson Technisch Laboratorium, Germany) approximately at an average rate of −1°C/min from 4°C to −30°C and then rapidly cooled from −30°C to −80°C at a maximum rate of −2.5°C/min. The frozen samples were thawed in NaCl 0.9% (Braun, Melsurgen, Germany) at room temperature for 7 min. After thawing, samples were washed once in NaCl 0.9% (Braun) for 5 min.
Decellularization
Dissected nerves were cleaned of fat and segments of 7 cm were used for decellularization. Decellularization was performed by serial incubations of zwitterionic sulfobetaines 10 and 16 (Sigma-Aldrich, Munich, Germany), nonionic detergent Triton X-100 (Sigma-Aldrich), hypertonic 1 M NaCl (Sigma-Aldrich), and 0.1 mg/mL Pulmozyme DNASE (Roche, Barcelona, Spain), followed by washes in 0.5 M Tris-EDTA buffer and ultrapure water at room temperature during 4 days.
DNA content analysis and DNA fragmentation assay
Native and decellularized samples were freeze dried. In brief, samples were packed, sealed, and frozen immediately at −20°C in Tyvek® bags. Freeze drying was conducted at −20°C using a pressure of 0.020 mbar for 22 hours. The final water content of the samples was <5%. Tissue (10 mg) was lysed with 0.5 mg/mL papain (Sigma-Aldrich) at 60°C during 3 hours. The lysate was used for DNA isolation with the DNeasy blood and tissue kit (Qiagen, Düsseldorf, Germany) following the manufacturer's instructions. DNA was quantified by Picogreen DNA quantification kit also following manufacturer's instructions (Invitrogen, Carlsbad, CA, USA) in a Tecan Spark multimode plate reader (Tecan, Männedorf, Switzerland) at 488 nm.
Preservation postdecellularization
Decellularized nerves were immersed in a cryopreservation solution (cryoprotectant) that was composed of trehalose (Life Sciences Advanced Technology) and HSA (Grifols), ratio 1:1 in RPMI without phenol (Gibco). Then, samples were frozen in an automatic controlled rate freezer (Cryoson 2010) as commented previously. Frozen samples were stored at −80°C and thawed as described earlier for downstream assays.
Histology
Samples were fixed in 4% paraformaldehyde (PFA) for 16 hours at 4°C, washed and paraffin embedded. Five microns sections were dewaxed, rehydrated, and antigen retrieval was performed. Slides were blocked and incubated with anti-laminin antibody (L9393; Sigma-Aldrich) at a dilution of 1/40 for 1 hours at room temperature. After several washes with phosphate-buffered saline (PBS), donkey anti-rabbit Cy3 IgG secondary antibody (Jackson Immunoresearch, Cambridge, United Kingdom) was incubated for 2 hours at room temperature at a dilution of 1/200 at 37°C. Masson's trichrome staining was performed in 5 μm sections as previously described. 15 Images were acquired with a fluorescence microscope (Olympus BX51) coupled with an Olympus DP76 camera for digital image acquisition. Pictures were analyzed with ImageJ software. 16
Indirect immunofluorescence
Samples were fixed in 4% PFA for 16 hours at 4°C, sucrose cryoprotected and embedded in Tissue Tek® OCT compound (Sakura, Fleminweg, Netherlands). Then, 5 μm sections were cut, permeabilized, blocked, and incubated with Fluoromyelin red fluorescent myelin stain (F34651; Invitrogen) for 16 hours at 4°C at a dilution of 1/300. After several washes with PBS, slides were mounted.
Quantifications of collagen and sulfated glycosaminoglycans
Quantitative biochemical assays were performed to determine the concentration of total collagen and sulfated glycosaminoglycans (sGAG). Five milligrams of freeze dried samples were used for collagen quantification by the hydroxyproline-based total collagen kit (Quickzyme Biosciences, Leiden, Netherlands) following the manufacturer's instructions, whereas 10 mg of these dried samples were used to quantify sGAG using the Blyscan sulfated glycosaminoglycans assay (Biocolor, Ulster, United Kingdom), also following the manufacturer's instructions.
Enzyme-linked immunosorbent assay
Fresh homogenized samples were used for enzyme-linked immunosorbent assay (ELISA) assays at a concentration of 100 mg/mL. Samples were lysed in RIPA buffer (Sigma-Aldrich) supplemented with protease inhibitors (Cultek, Barcelona, Spain) in a Precellys evolution homogeneizer (Bertin Technologies, Montigny-le-Bretonneux, France). ELISA for laminin (LSBio, Seattle, WA, USA) and S100 (LSBio) were performed following the manufacturer's instructions. Absorbance was read in a Biotek plate reader (Biotek, Colmar, France) at 450 nm.
Biomechanical testing
The Uniaxial tension assay was carried out on an INSTROM 3366 electromechanical testing system (Instrom, Turin, Italy). Samples had a final length of 30 mm and the clamps were separated by a distance of 10 mm. The samples had an approximate diameter of 2 mm, and the grips were lined with sandpaper. The samples were held at 4 bar of compression. The test was assayed at room temperature with the following specified settings: 0.1 N of preload, 12 mm/min of constant strain rate, and 40% of sensibility to stop the measure. Young's modulus and tensile strength were measured. 17
In vitro cytotoxicity assay
3T3-J2 cells were seeded in 96-well plates at a density of 2 × 104 cells/cm2 and cultured in standard medium (Dulbecco's Modified Eagle medium [Invitrogen] with 10% FBS [Biowest, Nuaillé, France] and 1% of penicillin, streptomycin, and amphotericin B [antibiotic antimycotic solution; Sigma-Aldrich]) at 37°C and 5% of CO2. To evaluate the cytotoxicity of the cryoprotectant used for preservation postdecellularization, the standard medium was changed by the cryoprotectant after 24 hours. For comparison, trehalose (w/v; Sigma-Aldrich) and bovine serum albumin (w/v; Sigma-Aldrich) from powder preparations were diluted in standard medium and added to the cells. Twenty-four hours later, the WST-1 assay (Abcam, Cambridge, United Kingdom) was carried out following the manufacturer's recommendations. Plates were read at 450 nm with a reference wavelength of 680 nm in an absorbance plate reader (Biotek). Sodium dodecyl sulfate 0.01% (Sigma-Aldrich) was used as a positive cytotoxicity control and cells with standard DMEM medium were used as a negative control.
To evaluate the toxicity of the nerve extracts, nerves were thawed at room temperature with 0.9% NaCl (Braun) as mentioned before. Nerve extracts were processed according to ISO 10993 recommendations. 18 In brief, 0.2 mg/mL of thawed nerves were immersed in standard medium for 24 hours at 37°C. Cytotoxicity was tested as described earlier. Latex was used as a positive cytotoxicity control and cells with standard medium were used as a negative control following ISO 10993 guideline. 18 Calculations were performed according to ISO 10993 guidelines. 18
Viability assay
3T3-J2 cells were detached from the culture plates and viability was measured with 4′,6-diamidino-2-phenylindole (DAPI; 200 μg/mL; Sigma-Aldrich) by flow cytometry in a MACSQuant cytometer (Mylteni Biotech, MA, Spain). Histograms were analyzed with MACSQuantify software (Mylteni Biotech). In parallel, the same detached cells were frozen with either DMSO 10% (WAK-Chemi medical GmbH) in FCS (Biowest) in a Mr. Frosty freezing container (Thermo Fisher Scientific; Life Technologies, Carlsbad, CA, USA), or in cryoprotectant in an automatic controlled rate freezer (Cryoson 2010) as described earlier. Cells were thawed at 37°C and viability was assessed by flow cytometry as commented.
Statistical analysis
Experiments were performed at least in triplicate. The two-tailed Student's t-test was used for comparisons and p-values <0.05 were considered statistically significant. All results are presented as the mean ± standard error (MD ± SE).
Results
Native nerves maintained histological structure regardless the preservation protocol but cell morphology changed
To elucidate the optimal preservation of native nerves before the decellularization step, we compared the structure of native control nerves with native nerves preserved for 7 days at 4°C with RPMI, DMSO- and trehalose-frozen nerves using Masson's trichrome staining.
Masson's trichrome staining showed intact collagen structure in all conditions (Fig. 1a). However, cell morphology changed among treatments. Cells in native DMSO-frozen nerves were smaller and showed a reduced cytoplasm in comparison with native nerves before preservation. In contrast, most cells of native trehalose-frozen nerves showed an expanded cytoplasm. Some cells of native nerves preserved at 4°C with RPMI disappeared, whereas the rest showed a decreased cell volume (Fig. 1a).
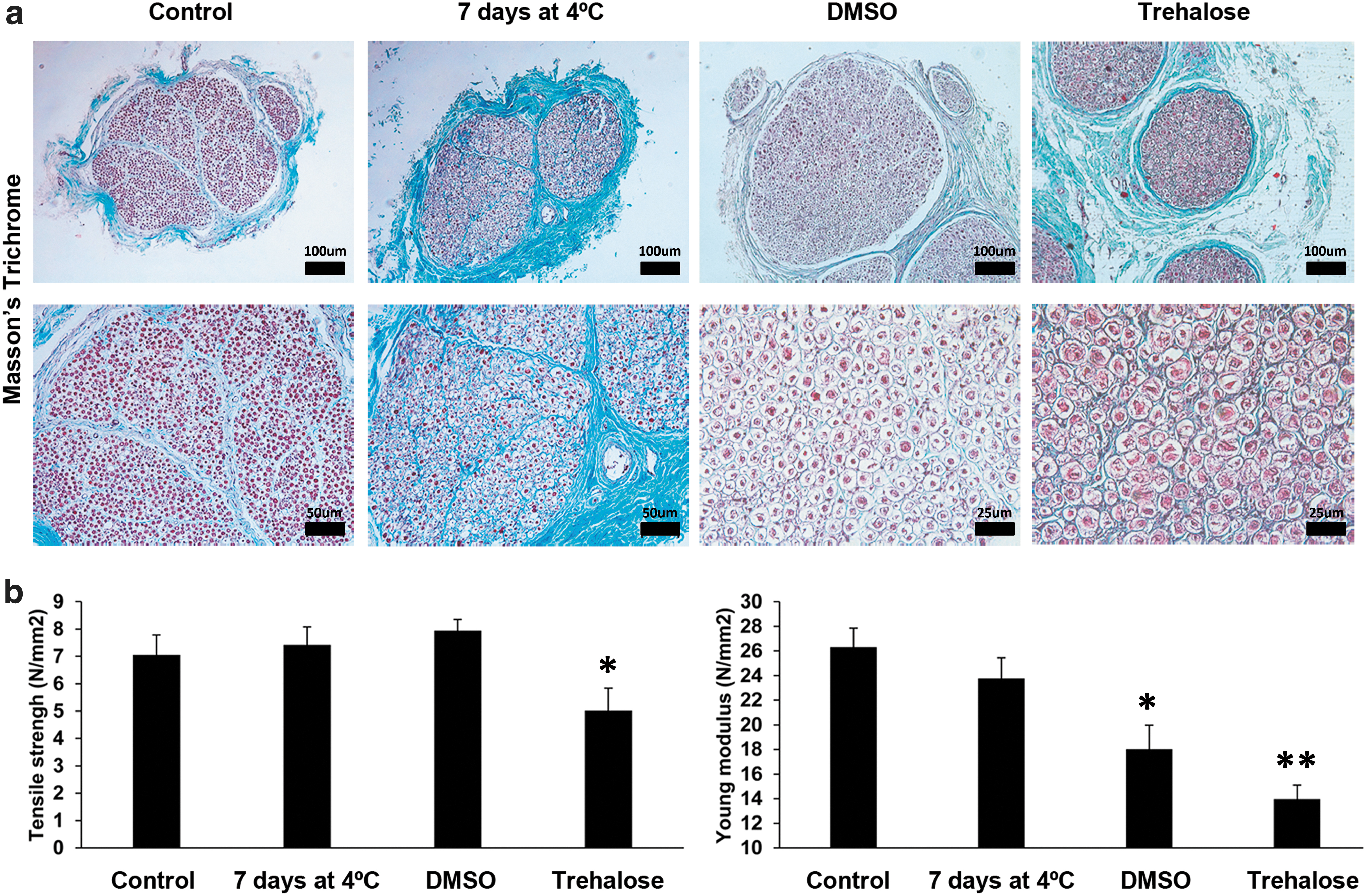
Different preservation strategies for the native nerves.
Preservation of native nerves at 4°C maintained biomechanical properties
To determine whether the preservation process of the native nerves could affect the biomechanical properties, we analyzed the tensile strength and the Young's modulus of the matrix as measures of resistance and elasticity, respectively.
Biomechanical properties of both native DMSO- or trehalose-frozen nerves showed decreased Young's modulus (Fig. 1b). Trehalose-frozen nerves also showed decreased tensile strength. However, native nerves preserved up to 7 days at 4°C maintained their biomechanical properties (Fig. 1b). On the strength of these results, only native nerves preserved at 4°C were considered for decellularization.
Preservation of native nerves up to 7 days at 4°C maintained ECM composition and endoneurial tube structure
However, before subjecting the nerves preserved at 4°C to decellularization, we investigated whether storage for up to 7 days could maintain ECM composition and endoneurial tube structure. To that end, we compared the concentrations of some key ECM proteins between native control nerves and native nerves preserved up to 7 days at 4°C by ELISA or biochemical quantifications and performed immunostaining for laminin.
Endoneurial tube structure was well preserved up to day 7 as demonstrated by laminin immunostaining (Fig. 2a). Using an ELISA assay we showed that there were no differences regarding laminin concentration between controls and preserved nerves (Fig. 2b). Moreover, biochemical quantifications demonstrated similar concentrations of total collagen and sGAG proteins between both conditions (Fig. 2b).
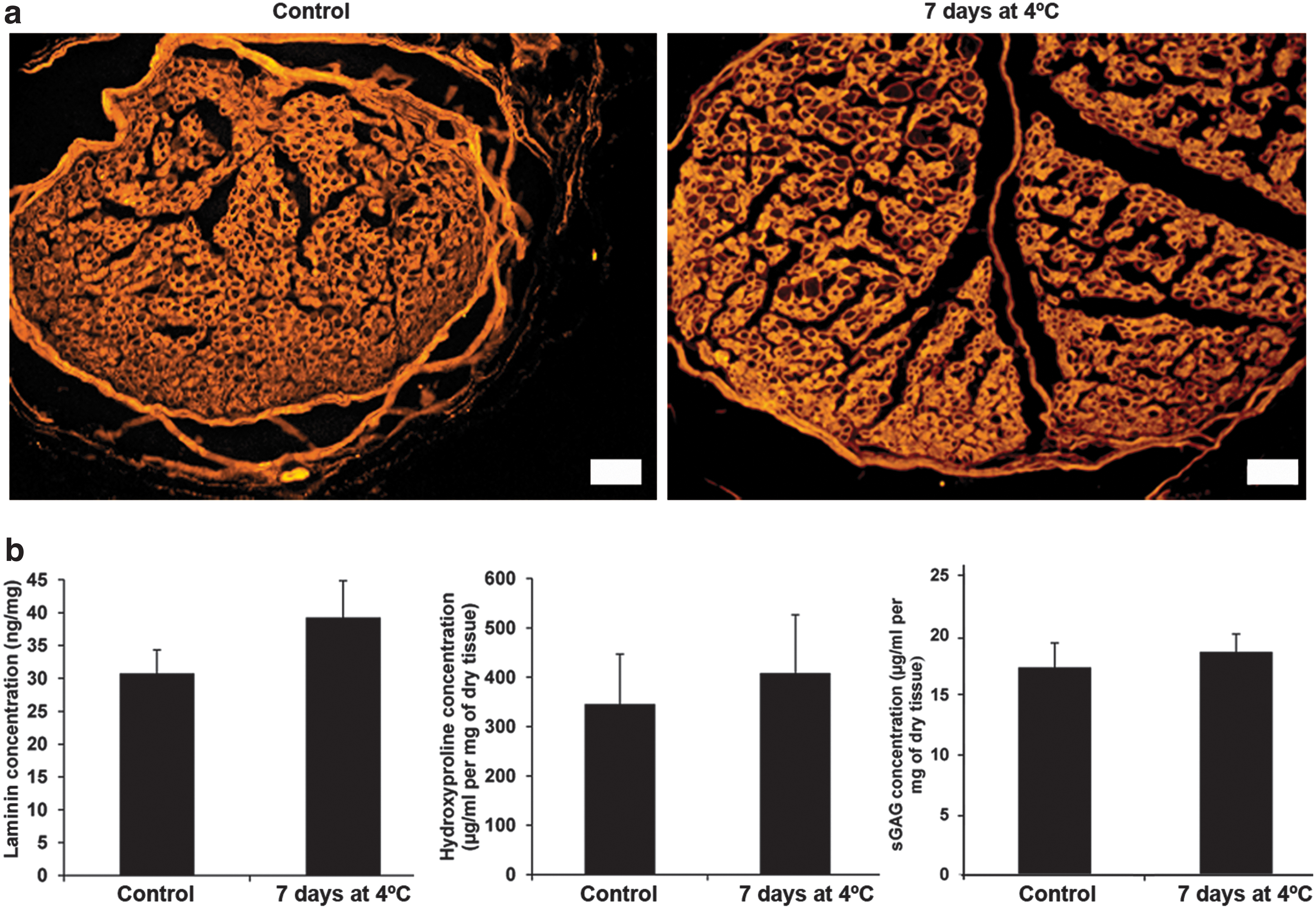
Maintenance of structure and composition of native nerve preserved at 4°C.
Decellularization process removed efficiently cell remnants
So, we used nerves preserved at 4°C to determine the efficiency of decellularization. We quantified the DNA concentration, performed DAPI staining for nuclear remnants examination, immunofluorescence for myelin and ELISA assays for S100 protein, a Schwann cells protein.
The concentration of DNA decreased below the threshold of 50 ng/mg of dry tissue in the decellularized samples (Fig. 3a), 19 which correlated with the absence of nuclear staining (Fig. 3b). In decellularized samples ELISA for S100 showed practically null expression (Fig. 3a). Moreover, myelin was not detected (Fig. 3b).
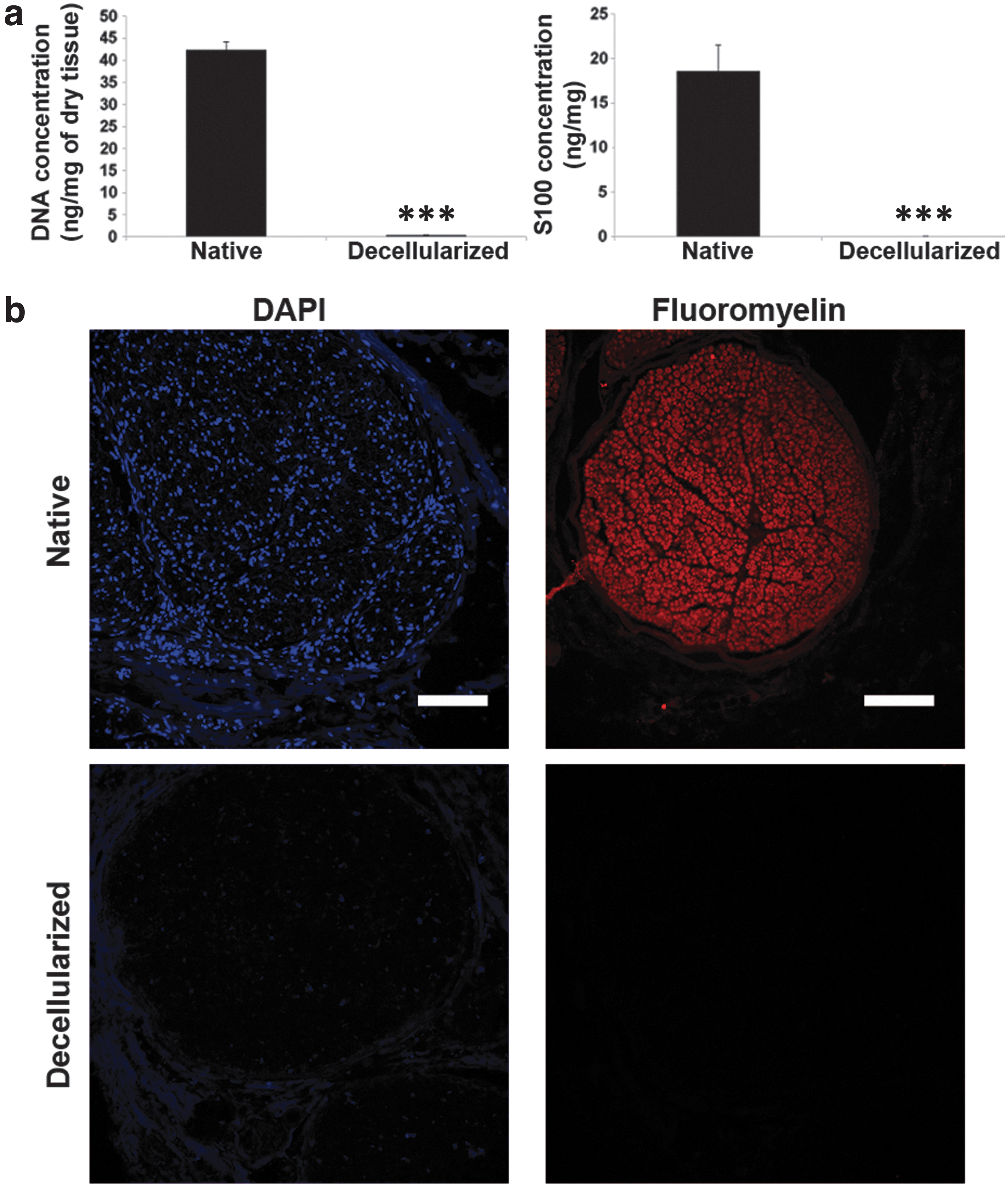
Decellularization efficiency.
Decellularized nerve extracts are not cytotoxic, whereas direct application of the cryoprotectant generates cytotoxicity on cells
We assayed the potential cytotoxicity for both the cryoprotectant and the extracts of decellularized nerves that were frozen with it. To do so, we followed the ISO 10993 guidelines, in which cellular viabilities <70% are considered toxic. 18
The direct application of the cryoprotectant on 3T3 cultures highly impaired the viability of the cells and changed the morphology, as observed in the positive controls for cytotoxicity (Fig. 4). Since the cryoprotectant was comprised of trehalose and HSA in plain RPMI medium, we asked whether the toxicity could be caused by the absence of nutrients. So, the same concentrations of trehalose and bovine serum albumin were diluted from powder preparations in 3T3 standard medium and the viability was highly improved (Fig. 4), indicating that the cytotoxic effects of the cryoprotectant were mediated mostly by nutrient deprivation.
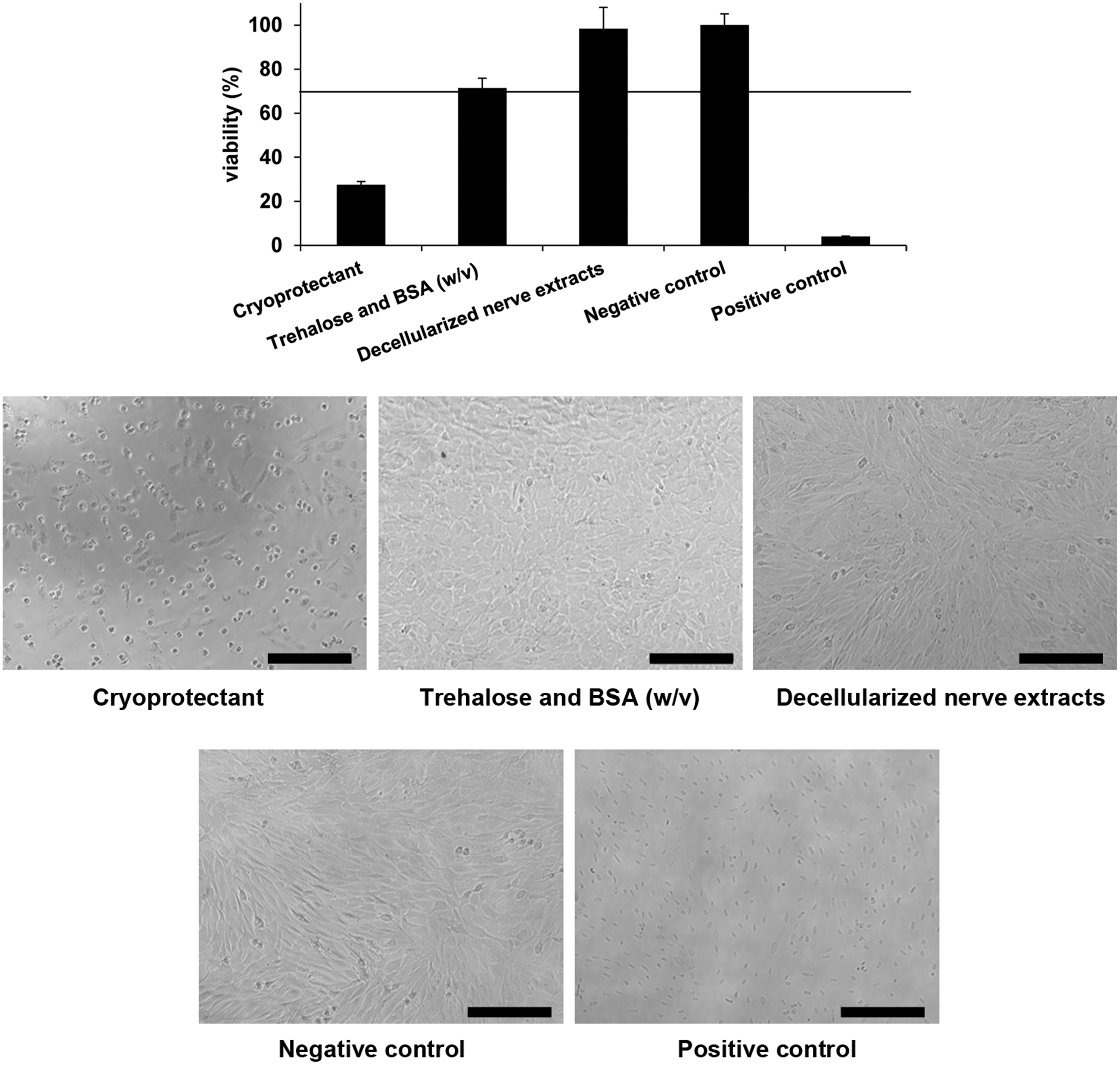
Cytotoxicity on 3T3 cells. Upper graph shows WST-1 assay results expressed as percentage of viability. Line shows the threshold below the reduction in the viability is considered cytotoxic (70%). Pictures show cell morphology in the different situations. Results are presented as mean ± SE from three independent experiments. Scale bar = 100 μm.
The evaluation of the nerve extracts demonstrated good preservation of cell viability and the cells presented similar morphology to the negative control of cytotoxicity (Fig. 4). This indicated that there were not toxic chemical remnants entrapped in the cryopreserved matrix after the thawing process.
Decellularized cryopreserved nerve maintained the structure, mechanical properties, and ECM composition
Furthermore, we probed the efficiency of the preservation protocol using the cryoprotectant within the decellularized nerves. We compared the control native nerves, the decellularized nerves, and the decellularized cryoprotected nerves for structure using histology for collagen and laminin. We also quantified ECM composition using ELISA as well as performed biochemical assays for laminin, total collagen, and sGAG. Moreover, we analyzed the mechanical properties such as Young's modulus and tensile strength.
Histological analysis for the structure of collagen and laminin did not demonstrate differences between controls, decellularized samples, and decellularized cryoprotected nerves, showing that endoneurial tube structure was well preserved between treatments (Fig. 5). Moreover, protein quantifications of collagen, laminin, and sGAG were maintained when the decellularized cryoprotected nerves were compared with native control nerves (Fig. 6a). Biomechanical testing did not show differences between controls and the decellularized cryoprotected nerves regarding resistance and elasticity (Fig. 6b).
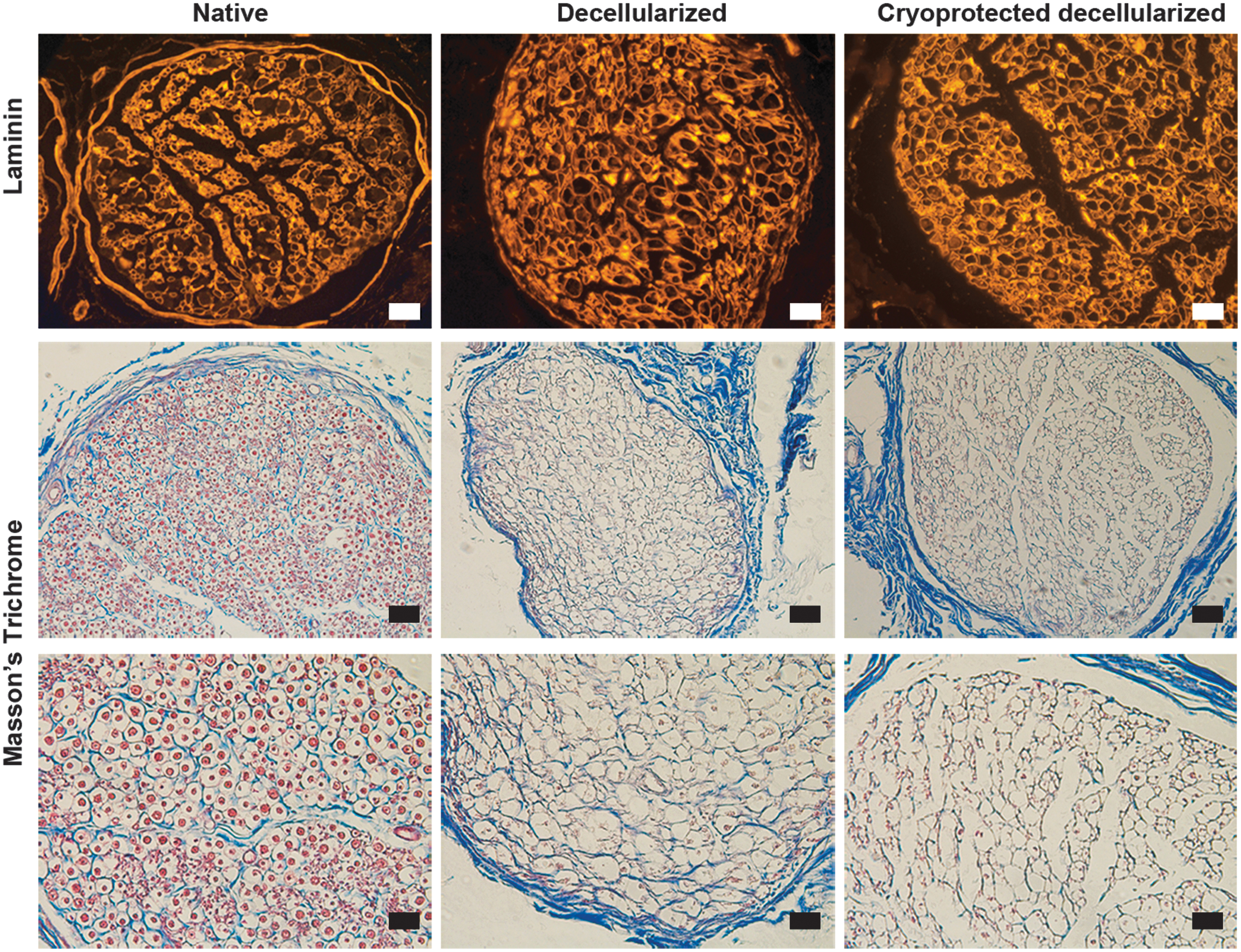
Maintenance of the structure of nerve matrix. Upper panels show laminin immunohistochemistry for native, decellularized, and cryoprotected decellularized nerve matrix. Middle and lower panels show Masson's trichrome staining. Scale bar upper and middle panels = 50 μm. Scale bar lower panel = 25 μm.
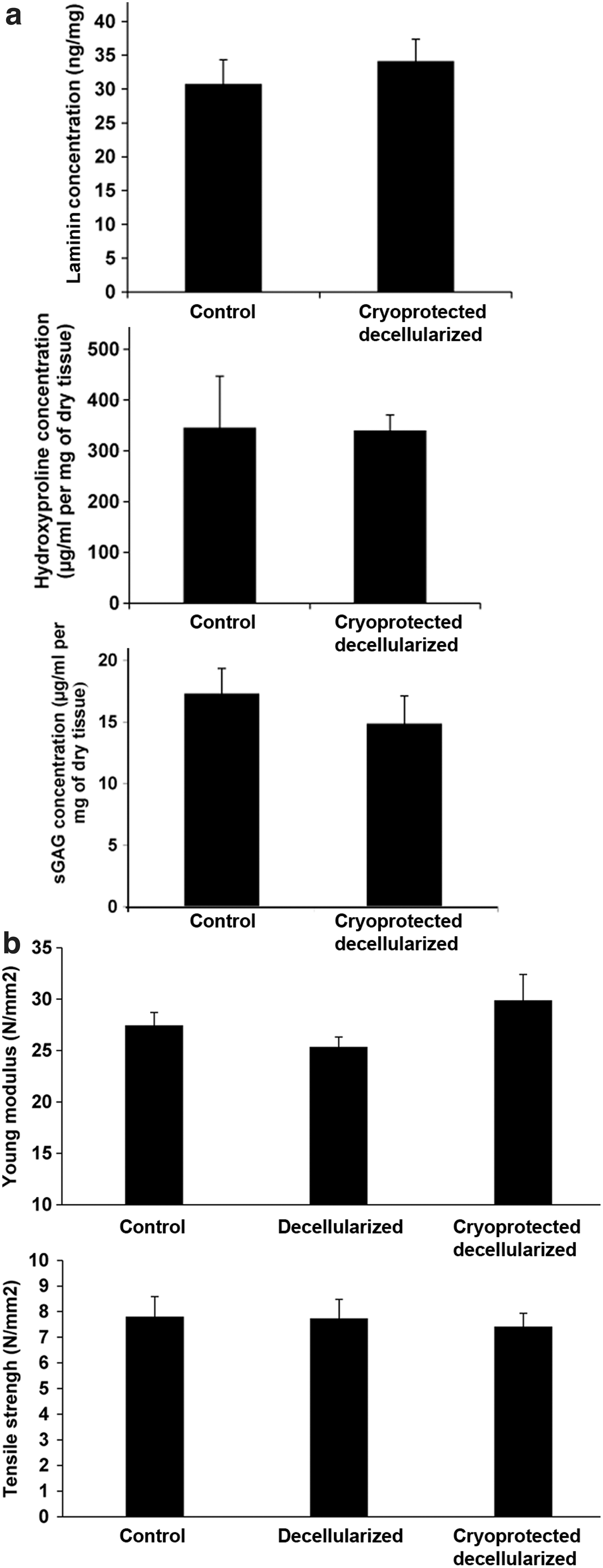
Maintenance of ECM composition and biomechanical properties.
The cryoprotectant maintained cell viability after freezing
We tested the cryoprotectant to maintain the viability of 3T3 cells after thawing in comparison with standard freezing medium, as a first approach to ascertain whether this cryoprotectant could be used to freeze 3T3 cells seeded on nerve matrix for future tissue engineering applications, since the nerve matrix would be frozen with the cryoprotectant in these applications. We froze the cells with 10% DMSO in FCS using the standard protocol of −1°C/min for freezing or with the cryoprotectant using an automatic controlled rate freezer as noted in the Materials and Methods section. We evaluated the cell viability before freezing and after thawing using flow cytometry. The results showed higher cell viability before freezing and after thawing. Moreover, there were no differences between both freezing protocols with either DMSO cryopreservation solution or the cryoprotectant formulation (Fig. 7).
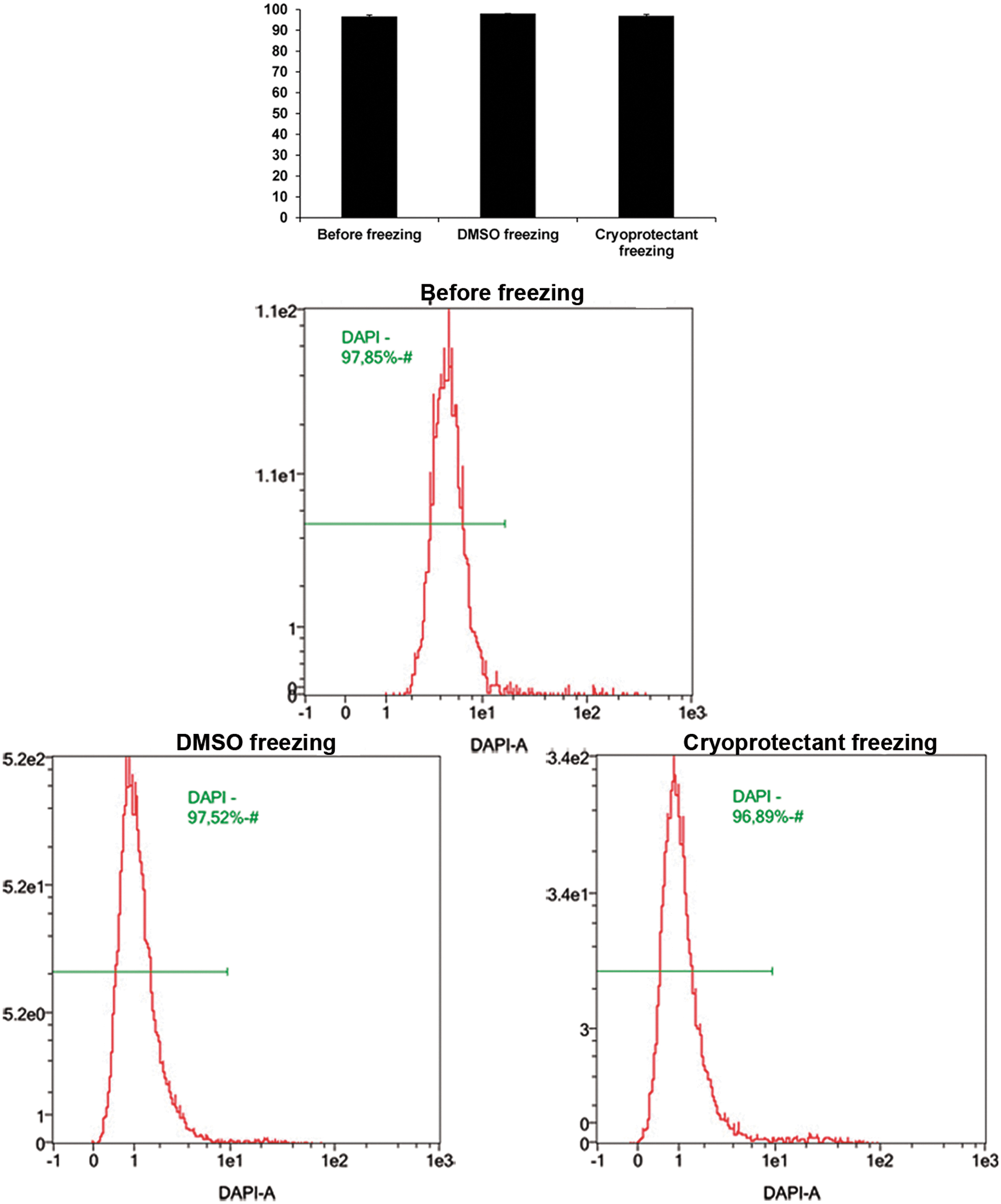
Comparison of the viability of 3T3 cells by flow cytometry before freezing, frozen with a standard protocol (DMSO freezing) and frozen with the optimized protocol plus the cryoprotectant (cryoprotectant freezing). Results are presented as MD ± SE from three independent experiments. Statistical analysis was performed using two-tailed Student's t-tests.
Discussion
The use of biological scaffolds is challenged by their shelf life, limiting their storage and availability. Preservation at low temperatures makes the biological scaffolds accessible, overcoming these drawbacks. Thus, the possibility to replace, just after an injury, the damaged native nerve by a ready-to-use decellularized nerve would enhance the quality of life of thousands of people.
In this study, we tried an integrated approach to the preservation of nerve pre- and postdecellularization. Our results showed that the properties of the native nerve are better preserved up to 7 days at 4°C than when frozen at −80°C with either trehalose or DMSO. Trehalose is used as an nonpenetrating cryopreservative, 20 so it could not enter through the native tissue to avoid crystal formation, hampering the biomechanical properties. Cell cytoplasm expansion with this treatment could also result in cell damage by ice formation. In contrast, DMSO is a penetrating cryoprotective agent, 20 so it could enter through the native tissue to avoid ice crystal formation. However, our data showed that DMSO had a negative impact in Young's modulus, increasing the elasticity of the native nerve. Recent findings suggest that DMSO induces dehydration of lipid bilayers, impacting their structures. 21 Since Schwann cells that form the myelin sheath are basically composed of multiple lipid bilayers, 22 this could explain the shrinkage of cell morphology after the thawing process of the sample.
Predecellularization preservation up to 7 days at 4°C maintained the biomechanical properties, the ECM composition and structure, although cell loss in the native nerve was observed. Far from being a disadvantage, this could favor matrix decellularization since, in this case, the ECM is maintained. In fact, some nerve decellularization protocols are based on cold (4°C) long-term storage 2 or cell apoptosis induction by cytotoxic substances to promote decellularization. 23 However, decellularization by cold long-term storage does not completely remove cell remnants and elicits an immune response. 2 Also, decellularization by apoptosis induction could imply a risk in generating neuron toxicity in the recipient. 24
After successful nerve decellularization, in terms of elimination of cell rests and ECM maintenance, we use a cryoprotectant composed of trehalose and HSA for preservation at −80°C for final storage. DMSO was not considered for the formulation of the postdecellularization cryopreservation solution because of its well-documented properties of blocking nerve conduction, 25 so we surmised that it could entail risks after nerve matrix transplantation.
Our data showed that nerve matrix properties, such as biomechanics, ECM composition, and structural integrity, were well preserved after cryopreservation with trehalose and HSA, the main components of the cryoprotectant. In this study, the detergents used for the decellularization treatment, permeabilized the matrix. This could allow the penetration of trehalose through the tissue, avoiding formation of ice crystals during the controlled freezing process and explaining the maintenance of the characteristics of the nerve matrix.
The election of trehalose for the cryoprotectant was based on its properties. Trehalose is a naturally produced disaccharide, which in combination with soluble proteins in nature avoids crystallization, allowing the protection of cell structures and proteins against damage in low temperature environments. 26 Previously, trehalose has been used to freeze alginate-encapsulated cells 27 and tissue-engineered epithelial sheets, 28 without exhibiting cytotoxicity, as we also demonstrated with 3T3 cells. We showed that the cryoprotectant exerted cytotoxicity mostly by cell nutrient deprivation. However, it should be considered that the toxicity that can be caused by a cryopreservation solution in culture conditions has different implications than the cytotoxicity caused by the scaffold. For this reason, using ISO 10993 recommendations 18 we tested the nerve extracts after the thawing process, demonstrating the nerve matrices were cytocompatible. This demonstrated that any toxic residual reagent was eluted from the nerve matrix. The cryoprotectant yielded higher cell viability after thawing 3T3 cells that were frozen alone, without being seeded in the decellularized nerve matrix. Therefore, this new freezing protocol would be of interest in the case that the nerve matrix would be used for further bioengineering applications, because it is likely to maintain the viability of the cells seeded and frozen for the nerve matrix.
In addition, the storage temperature of the decellularized nerve matrix was −80°C, avoiding the use of nitrogen liquid, which increases the risk of cross-contamination and safety hazards. Storage at −80°C would simplify more handling, storage, and shipping than using liquid nitrogen. This would decrease manufacturing costs, making the production more cost-effective.
In this study, we demonstrated the production of “off-the-shelf” decellularized nerves, using methods that preserved the properties of the matrix before decellularization and postdecellularization, aiming for future clinical application for regenerative medicine purposes.
Footnotes
Authors' Contributions
N.N.-N. designed, performed experiments, analyzed data, wrote the article, and provided final approval of the article for publication. P.L.-C., R.P.C.-M., O.F., and A.V. critically revised the article. TC provided technical assistance and critically revised the article. E.C.-C. and S.B. performed experiments and analyzed data. E.U. and X.N. analyzed data.
Acknowledgments
We thank the donors and their families that with their altruistic act of donation allowed us to perform this study. We also thank the transplant coordinators, donor center, and recovery teams for their efforts that make it possible to obtain the samples used in this study, and T. Van Eeckhout for his assistance in the final editing and preparation of this article. Finally, we thank Lola Mulero from the histology service of the Center of Regenerative Medicine of Barcelona, for her kind counseling.
Author Disclosure Statement
No conflicting financial interests exist.
Funding Information
This study has been partially supported by grant from TERCEL (RD16/0011/0035) and CIBERNED (CB06/05/1105) from the Instituto de Salud Carlos III (Madrid, Spain), and internal competitive grant from BST (Ref I.2017.056).
Supplementary Material
Graphical abstract