
Abstract
Animal cloning is an important technique used to produce clones from valuable farm animals, to rescue animals in risk of extinction, and for producing transgenic animals. The objective of this work was to evaluate the effects of refrigeration on bovine ear skin as a strategy to transport biological material for long periods of time to isolate viable fibroblasts. Ears from eight cows were collected after death and stored for 30 days at 5°C. On days 0, 2, 4, 7, 14, 21, and 30, skin biopsies were cultured in vitro for fibroblast isolation. The time for first fibroblast outgrowth, time to reach 100% confluence. and cell concentration before freezing were observed for each period. In addition, plasma membrane integrity, cell apoptosis, and necrosis in cells were evaluated through fluorescent colorant combination in a flow cytometer from all periods after thawing. Fibroblasts obtained after 30 days of storage, considered a critical period, were tested for embryo production using nuclear transfer (NT) with micromanipulators. All time points allowed for cell culture. The time of cell growth onset was longer in samples refrigerated for 14, 21, and 30 days. The time to reach confluence also increased with longer refrigeration periods. Cells from day 0 reached confluence in 24 ± 2 days, while day 30 cells took 31 ± 0 days. Cell concentration and viability dropped with increased storage time and freezing/thawing, respectively. It was found that a long period of sample storage results in cell damage, making cultivation more difficult and decreasing cell viability post-thawing and cell concentration. However, when cells from day 30 were used as nuclei donors in NT, a 26.05% blastocyst rate after 7 days in culture was obtained. In conclusion, refrigeration at 5°C was shown to be efficient in maintaining viable tissue for up to 30 days, and fibroblasts isolated can be used for cloned embryo production.
Introduction
It is known that epigenetics plays a major role in nuclear reprogramming, and mechanisms such as DNA methylation and histone acetylation are currently on the radar of a great number of researchers due to their importance in nuclear transfer (NT) success. 1 Most experiments aiming at epigenetic improvement have focused on the nuclei donor cell, 2 showing that the cell selected to be the nuclei donor is of major importance, because it carries the desired DNA.
Genome integrity is essential for successful cloning of animals. 3 Thus, the quality of nuclear donor cells, including their viability and proliferation in culture, ensures their genomic integrity and enhances the success rate of full-term development of animals in a cloning experiment. 4 The isolation and conservation of viable cells is the first step before using preserved material in an NT technique, aiming for the multiplication of farm or wild animals. Cloning by NT using somatic cells as nuclei donors allows the isolation of cell lines to become an alternative to the conservation of genetically valuable animals. 5
The competence of blastocysts produced by NT has been demonstrated initially by the production of live animals 6 and secondly by the derivation of embryonic stem cells from the embryo's internal cell mass. 7 These observations provided definitive proof that a somatic cell nucleus may be reprogrammed to a pluripotent state by the oocyte's cytoplasmic factors, and that the reprogrammed nucleus may develop into a full individual.
Starting from the tissue sample removal from the donor animal, the survival and proliferation of cells drop due to pH changes, the accumulation of toxins such as oxygen reactive species, 8 and the progressive RNA degradation, consequently reducing the gene expression rate and leading to tissue death and the unfeasibility of the cultivation of cells present in the tissue. 9 Therefore, the use of low temperatures is a strategy for longer tissue viability conservation. Somatic cell conservation from different species has been performed using cooling at low temperatures of 4°C–6°C,5,8,10–12 slow freezing,13,14 or vitrification. 15 A critical aspect for the establishment of primary cellular lineages is the period between biopsy collection and sample processing to initiate cultivation. In continental countries, animals may be in faraway properties or farms with difficult access, which may greatly extend this period.
In this research, we have evaluated if bovine skin fragments refrigerated at 5°C up to 30 days postmortem could still be used for fibroblast isolation and culture, verify cellular proliferation patterns in different periods of time, and determined if these cells could be successfully used in the production of cloned embryos.
Materials and Methods
Experimental design
The experiment was delineated in randomized blocks with seven treatments (refrigeration time points) and eight blocks (ears). The effects of treatments over outgrowth time, confluence, concentration, cell viability, apoptosis, and necrosis post-thawing were evaluated by variance analyses (ANOVA) and comparison of media by Tukey's test with significance level of p < 0.05.
Ethics and animal welfare
All ears were excised from abattoir cattle or animals that died of natural causes in nearby farms, so no animal was slaughtered for this experiment, creating no ethical or moral barriers. All experimental procedures were approved by the Ethics Committee in Animal Use at Embrapa Cerrados (protocol no. 578-2886-2/2017).
Skin sample collection
For this experiment, ears of 6 Nelore bovine females, 24 months of age, from the same farm were collected at the moment of slaughter. Ears of another 2 Gyr cows over 15 years of age were also collected after death by natural causes in local farms. All samples were transported to the laboratory in a container with chemical ice and a thermometer that marked 5°C. Transportation lasted around 1 hour. In the laboratory, ears were trichotomized, washed five times with neutral detergent, rinsed with water, dried with sterilized tissue paper, sprayed with 70% alcohol, and left to dry completely before being stored in individual sterilized Ziplock®-type bags. All ears were stored whole to reduce open tissue exposure and kept in a temperature-controlled refrigerator at 5°C up to 30 days. On days 0 (day of death), 2, 4, 7, 14, 21, and 30 (named D0 through D30), cell cultivation was conducted.
Fibroblast isolation and culture
A small piece of each ear (3 × 3 cm) was cut off, washed with neutral detergent, dried with sterile tissue paper, sprayed with 70% alcohol, and left to dry in a sterilized petri dish. This second round of cleaning had the purpose of reducing contamination due to manipulation. The sample was then taken to the laminar flow hood where, with scalpel and tweezers, the skin was separated from the cartilage and 9 small pieces of skin (0.5 × 0.5 cm) were fabricated and placed on the bottom of a cell cultivation dish (35 × 10 mm, Eppendorf®). Three dishes were produced per animal, per time point. After waiting for 10 minutes so that the skin fragments could dry and attach to the bottom of the dish, the dish contents were then completed with 2.5 mL of Dulbecco's modified Eagle's medium (DMEM; Invitrogen Life Science, Rockville, MD), with 10% of bovine fetal serum and penicillin and streptomycin as antibiotics. 16 Dishes were observed every 2 days to locate and register initial cell outgrowth, identify and control possible contaminations, observe how long it took for cells to reach confluence and ultimately freeze those cells, and calculate cell concentration at the time of freezing. DMEM was changed for nutrient renovation every 4 days when no contamination was present and every 2 days when contamination was detected.
After explant removal, when cells reached around 70%–90% confluence, they were transferred to a 25 cm2 (50 mL) flask (Biofil®). When the cells reached 100% confluence inside, the flasks were analyzed and frozen.
The cellular solution in the 1.5 mL tubes was used to calculate cell concentration on a Neubauer chamber before freezing. In total, 64 squares were counted in each sample. The result was divided by 4 and the resulting number multiplied by 10 to obtain cell concentration per mL. 17
Flow cytometry analyses
For this analysis, an AMINS FlowSight Image Cytometer (Amnis Corp., Seattle, WA) was used. The acquisition software used was INSPIRE V6.1 and the analysis software was IDEAS V5.0.
Plasma membrane integrity, cell apoptosis, and necrosis were evaluated through fluorescent colorant combination in cells from all periods after thawing. The Dead Cell Apoptosis kit (AlexaFluor® 488 Annexin V; Molecular Probes, Inc., Eugene, OR, EUA) (A), propidium iodide (PI), and nuclear colorant Hoechst 33342 (bisBenzemide H33342 trihydrochloride—to eliminate possible debris and other grime) were the colorants of choice. Viable cells were registered as A− PI−, as they did not change color with any of the probes. Cells that were colored with AlexaFluor (A+) were at initial apoptosis, cells that were colored with both propidium iodide and AlexaFluor were at the final apoptosis stage and initial necrosis state and were identified as An+ PI+, and cells that were at final necrosis estate colored with propidium iodide and were identified as PI+.
Fibroblasts from all periods were evaluated to investigate possible differences in cell viability after thawing. Three repetitions from each sample were analyzed. Samples were brought to the laboratory frozen inside a cryogen canister. They were thawed in a 37°C water bath for 30 seconds. Cells were then placed in 1.5 mL Eppendorf tubes and centrifuged for 4 minutes at 150 g. After centrifugation, excessive supernatant was discarded and samples (20 μL) were incubated for 15–30 minutes at 37°C in a work solution (20 μL) prepared according to the manufacturer's instructions.
All samples were divided in two, forming one control group and one treatment group for each sample analyzed. Control groups had 10,000 single cells evaluated and were not treated with fluorescent probes. Treatment groups had 40,000 single cells that were analyzed after being treated with fluorescent probes. Images were obtained with an objective at 20 × magnification.
For flow cytometry, a specific gate was created with the following coordinates: X axis (area) = 155 to 1944 and Y axis (aspect ratio) = 0.371 to 0.997 for fibroblast only selection.
AlexaFluor (A) emissions were caught through channel 2 (505–560 nm) and excited using 488 nm at 30 mW laser. Propidium iodide (PI) emissions were caught in channel 4 (595–642 nm) and excited with a 488 at 30 mW laser. Signals emitted from Hoechst 33342 were detected at channel 7 (435–505 nm) after laser excitement of 405 nm at 45 mW. SSC laser was used at 10 mW.
Cellular freezing
When in 100% confluence inside 25 cm2 flasks, cells were detached from the bottom of the dish with 0.25% Trypsin (Sigma-Aldrich) and incubated for 6 minutes, transferred to 1.5 mL Eppendorf tubes and centrifuged for 6 minutes at 900 g. Once centrifuged, the supernatant was discarded, leaving only the cell pellet at the bottom of the tube. The pellet was then suspended with a solution composed of 90% DMEM and 10% Dimethyl Sulfoxide (Me2SO) as cryoprotectant. Once suspended and homogenized, the solution was then pulled into 0.25 mL identified straws and sealed. Straws were then stored for 24 hours in a −80°C freezer and then dipped into liquid nitrogen, and stored in liquid nitrogen canisters. 17
Somatic cell nuclear transfer
When in 100% confluence for ∼2 days (when cells had proliferated and covered completely the bottom of the cultivation dish), day 30 fibroblasts were used in the NT process as donor nuclei to investigate if they could be successfully used to produce cloned embryos. Cells from the second passage from all donor animals were used.
For the cloning process, a Narishige micromanipulator was used (model IM-9B; Narishige, East Meadow, NY) comporting a Nikon inverted microscope (model Ti-S; Nikon Instruments, Melville, NY). The protocol used in this experiment was according to Silva et al. 18
Oocytes from abattoir ovaries were obtained, selected according to cumulus cells and cytoplasm integrity and only degrees 1 and 2 oocytes were maturated for 18 hours. After the 18-hour mark, cumulus cells were removed by pipetting for 2 minutes after oocytes remained for 3 minutes inside the incubator in hyaluronidase. Oocytes that presented the first apparent polar body were used for enucleation. Using the first polar body as reference, both the polar body and metaphase plates were removed and one fibroblast was injected into the perivitelline space. For confirmation of metaphase plate removal, oocytes were stained with Hoechst 33342 (bisBenzemide H33342 trihydrochloride).
After reconstruction, karyoplast-cytoplast couplets were fused in a microslide fusion chamber containing stainless steel electrodes with a 1 mm gap between them. An electron cell fusion/activation system (Voltain EP-1; Cryologic, Blackburn, Victoria, Australia) was used in couplet fusion. The fusion solution used was a 0.3 M mannitol that filled the chamber, allowing the couplet to be placed between the two electrodes and electric shock was conducted appropriately. Karyoplast-cytoplast couplets were fused in two consecutive DC pulses (pulse, 140 volts; alignment frequency, 100 kHz; pulse time, 20 ls; and amplitude, 0.7–14 peak volts). After a minimum of 20 minutes, couplets were evaluated for fusion by microscopic examination. For chemical activation, 150 couplets were exposed for 5 minutes to 5 μM of ionomycin and for 4 hours in 1.9 mM of 6 dimethylaminopurine (6DMAP). Mature oocytes presenting the first polar body were activated at the same time as fused couplers to be used as parthenogenetic control. All activated couplets were cultured in synthetic oviduct fluid with 5% FBS until 7 days.
Statistical analyses
Statistical analysis was performed on Sigma Plot 12 software. ANOVA was employed to verify the effect of cooling time in cell parameters before and after sample cryopreservation. Mean comparison was verified by Tukey's test at 5%. The T test was applied in cloned and parthenogenetic embryo mean production comparison.
Results
Cell culture, time to confluence, and concentration
Fibroblast isolation from refrigerated bovine skin was possible at all time periods (D0 through D30), but a significant difference in initial cell outgrowth was observed (p < 0.05). The more distant to the day of death (D0), the longer it took for fibroblasts to outgrow skin explants. While day 0, 2, and 4 cells took around 4–5 days to start proliferating, day 7, 14, 21, and 30 cells took a significantly longer amount of time, with day 30 fibroblasts taking up to 35 days to start proliferating around skin explants (Table 1 and Fig. 1).

Difference between the different time points in bovine fibroblast outgrowth. Cells from earlier time points D0
Mean and Standard Deviation of Days Taken for Detection of Initial Cell Outgrowth, Time Needed for Cells to Reach Confluence, and Cell Concentration on Freezing Moment from Fibroblasts Cultivated in Different Time Points
Different letters in columma,b,c,d represent statistically significant differences (p < 0.05).
The same growing pattern appeared regarding the amount of time it took for cells to reach 100% confluence. Cells from all time points reached 100% confluence; however, cells isolated in posterior periods of sample refrigeration took a larger amount of time to reach confluence when compared to cells isolated in earlier time points (p < 0.05; Table 1).
Cell concentration obtained from refrigerated skin biopsies at days 0 to 7 remained stable in the confluence time; however, cell concentration decreased sharply in the later periods of storage.
During cell cultivation, we observed that dishes produced in later time points presented a higher level of contamination. Up to day 7 postmortem, contamination was practically nonexistent, and when contamination was present, it was easy to control. No dishes were lost from days 0, 2, 4, and 7; however, dishes fabricated with biopsies from days 14, 21, and 30 postmortem were much harder to cultivate, given the fact that contamination was frequent. Many dishes were lost due to uncontrollable contamination, especially during days 21 and 30. At the end of the experiment, it was possible to cultivate, freeze, analyze, and use in NT cells from all time points, but cells from days 21 to 30 presented themselves as much harder to cultivate and reach 100% confluence than cells from earlier time points.
Flow cytometry analyses (viability)
Fibroblast viability percentage after freezing/thawing remained at ∼80% until day 14, suffering a large decrease on days 21 and 30 (Table 2 and Fig. 2). Besides that, there was also an increase of apoptosis and necrosis after thawing in cells obtained from skin biopsies refrigerated for days 14, 21, and 30 (Table 2 and Fig. 2). No differences were observed between animals of different breeds and ages regarding cell viability.
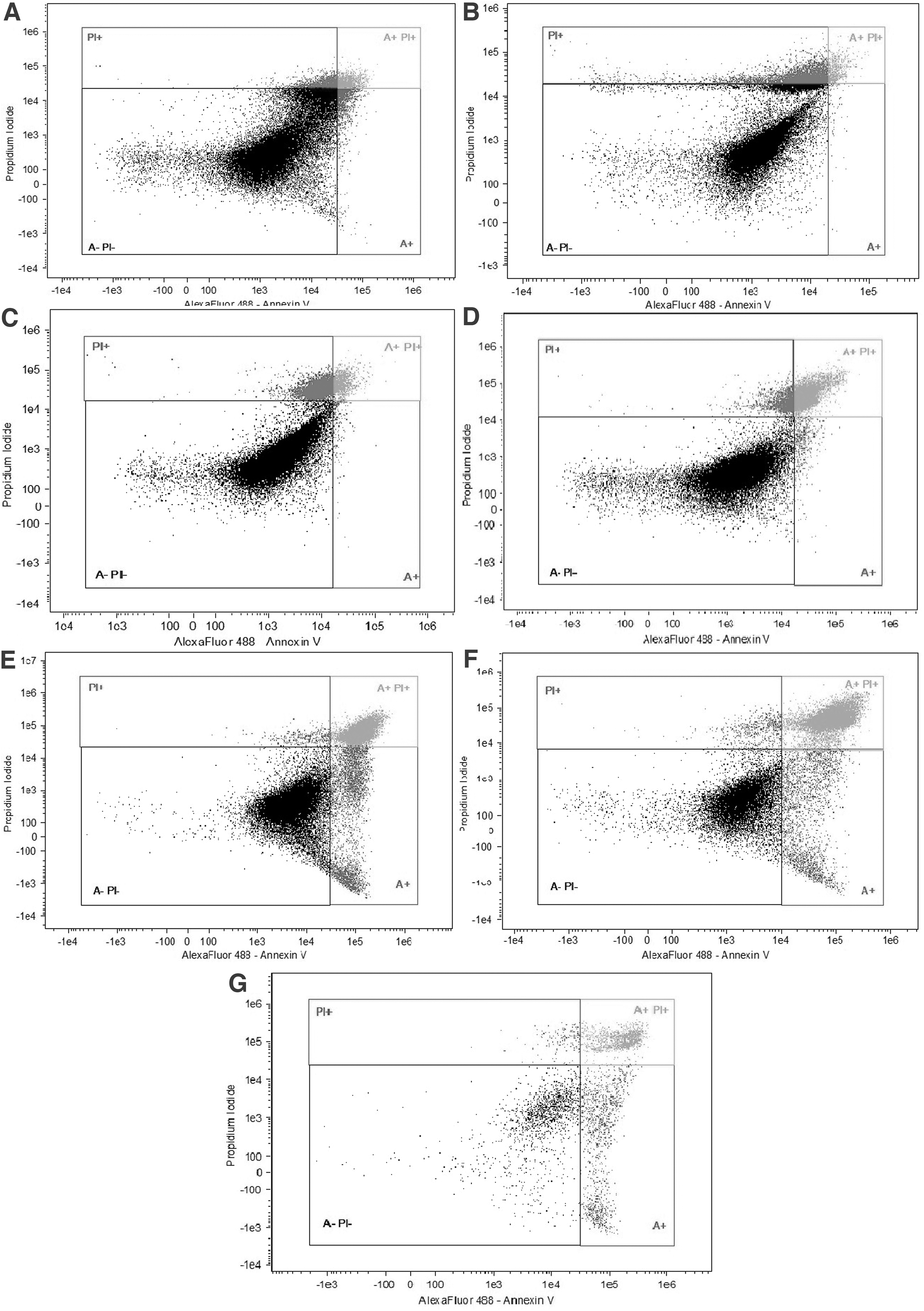
Comparison between day 0
Viability, Apoptosis, and Necrosis (Average ± Standard Error) Comparison Between Different Time Points Post-Thawing of Bovine Fibroblasts Obtained from Refrigerated Tissue for Up To 30 Days of Sample Storage
Different letters in the columma,b indicate statistical difference (p < 0.05).
Embryo production
Table 3 presents the results regarding electrofusion, cleavage, and blastocyst production after 7 days of culture in comparison to parthenogenetic embryos. A 26.05% blastocyst rate was achieved from all 119 fused couplets. While the p value for cleavage presented a statistical difference of 0.0491 (p < 0.05), day 30 NT blastocyst rate was similar to parthenogenetic embryo rate (p value was 0.0906, considering p > 0.05).
Electrofusion, Cleavage, and Blastocyst Rate Comparison Between D30 Cell Cloned and Parthenogenetic Embryos
Different letters in the columma,b indicate statistical difference (p < 0.05).
NT, nuclear transfer.
Discussion
The first step of bovine cloning by somatic cell nuclear transfer is the isolation and culture of the desired DNA. Nowadays, this is achieved through culture of skin fibroblasts from the ear or tail. Fibroblasts are currently the most used stem cells in bovine NT, given their easy culture, identification, conservation, and resistance. It was already observed that fibroblasts can be successfully used in NT to produce adult cloned animals, 19 and that their genome, even though already differentiated to a certain degree, could be reprogrammed into a pluripotent estate.
One of the biggest issues in farm animal cloning is that sometimes the animal dies or is housed in a property located far away from a laboratory, or the material can only be sent a few days after the animal's death.
Another problem faced in this reality is that because of the lack of an appropriate storage and shipping protocol, it is common for the laboratory to receive frozen samples, which makes cell cultivation difficult. On the other hand, sometimes the skin fragments arrive at the laboratory already in a state of putrefaction, which makes cultivation impossible. In this context, we have tested the conservation of skin tissue at 5°C for up to 30 days before fibroblast isolation and its use in NT.
In this study, we observed that cooling of the bovine ear skin at 5°C is an important strategy for transporting bovine tissue for long distances and recovery of viable cells up to 30 days after sample storage. However, the increase in cooling time interferes with the fibroblast proliferation patterns until freezing and cell viability after thawing from cryopreservation. The longer the ears remained inside the refrigerator, the harder it became to make skin explants, given their friable condition.
Despite the negative effects of skin cooling, viable fibroblasts were obtained from refrigerated biopsies for 30 consecutive days. Blastocysts were produced by NT, indicating that this methodology can be used in extreme cases of transport over long distances.
In this experiment, cellular isolation was performed at all skin storage times; however, the increase in cooling time had a direct impact on the time it took for onset of fibroblast growth. On days 0, 2, and 4, fibroblasts took 4 ± 0, 5.33 ± 1.5, and 4.33 ± 1.03 days, respectively, to start growing from biopsies, when in day 7, a significant increase in that time was observed (10.0 ± 1.78 days). Such an increase in time appeared in all subsequent periods, with special emphasis on day 30, which took 33.5 ± 1.5 days to observe fibroblast outgrowth. Silvestre et al., 11 Caputcu et al., 10 and Walcott and Singh 12 also observed bovine outgrowth of fibroblast, muscle, and cartilage cells, respectively, up to 12, 9, and 49 days postmortem.
A correlation between time of cooling and time to reach 100% confluence was also observed in our experiment. In earlier time points (days 0 and 2), cells needed 24 ± 2 and 28.0 ± 3.1 days to reach 100% confluence, while at days 4, 7, 14, 21, and 30, cells took significantly larger amounts of time. Walcott and Singh (2017) 12 observed that when compared with fresh biopsies, the biopsies fabricated from samples stored for 49 days presented a much slower growth rate, of only 16.7%. This increase in time to confluence may be due to the reduction of stem cells present in tissue with increased storage time.8,20 Cell survival and proliferation rate decrease may be attributed to pH changes and accumulation of toxins such as reactive oxygen species 8 and RNA progressive degradation, reducing the rate of gene expression and confluence. 9 During low temperature storage, a fast consumption of cellular energy reservoir occurs, primarily ATP followed by a gradual decline in total adenine nucleotide content and a consequent accumulation of lactate. 16
Regarding cell concentration, in our study, the long period of skin storage biopsies at 5°C caused a decrease in cell concentration at the time of freezing. Treatments D21 and D30 presented the lowest fibroblast concentration among groups. Moreover, in addition to reducing in quantity, later time point cells (days 14, 21, and 30) visually also presented a larger size when compared to day 0 to 7 cells. Such an increase in cell size may be explained by the increase in the number of halos in “older” cells. The presence of halos may have taken more space within the cell, making the cell occupy more space on the dish, giving the dish an appearance of confluence when the dish presented a significantly lower number of cells when compared to dishes that were in actual confluence from earlier time points, where cells showed a higher percentage of viable cells with consequently less apoptotic cells. Halos consist of structures that signal the onset of apoptosis and are formed after the separation of the nucleus from the cytoplasm.8,12 This observation was also made by Walcott and Singh 12 on their experiment with bovine ear skin explants.
We also have observed that post-thawing viability rates suffered a significant decrease in later storage time, dropping from around 78.66% ± 4.23% on days 0, 2, 4, 7, and 14 to 33.7% ± 4.3% on day 21 and 28.72% ± 4.81% on day 30. This abrupt drop in cell viability may be due to interference of long periods of tissue refrigeration that compromised cell metabolism and the growth pattern. In addition, we observed an increase in cell apoptosis related to longer ear skin storage periods.
Studies with other species achieved similar results. Okonkwo and Singh (2014) 5 observed outgrowth around goat skin explants up to 41 days postmortem, and reported that earlier time point cells took 4 days to outgrow, while later time point (days 37 and 41) cells took 25 and 32 days, respectively. Singh and Ma 21 achieved successful sheep outgrowth up to 65 days and Neta et al. 8 showed collared peccary fibroblast outgrowth up to 30 days without nutrient media. Silvestre et al. 22 verified that rabbit and pig fibroblasts could be obtained up to 10 and 14 days, respectively. As to cell viability, Ge et al. 23 also observed a drop in fibroblast viability with increased storage time with pig skin fibroblasts cultured up to 15 days postmortem, which presented with only 20% viability.
Neta et al. 8 also observed this phenomenon. Although all these studies demonstrate that it is possible to obtain fibroblasts after long periods of skin cooling, none of them has demonstrated the viability of these cells using the NT technique.
Moreover, we observed in this study that contamination of biopsies was more prevalent in the ears refrigerated for 21 and 30 days, particularly given the fact that even inside the refrigerator, ears started to decompose, as cold temperatures slowed down, but did not stop autolysis.
Despite the negative effects of long cooling periods, fibroblasts obtained from 30 consecutive day cooling biopsies allowed 26.05% of cloned blastocyst production, indicating that this methodology can be used in extreme cases of long-distance transportation. This blastocyst rate was similar to other articles that used fibroblasts obtained from fresh biopsies.17,19,24
We conclude that low temperatures of 5°C have been shown to be efficient in maintaining viable tissue for up to 30 days. However, significant changes in cell attachment and proliferation, concentration until freezing, and viability after cryopreservation can be observed the longer the tissue is stored. Despite such obstacles, it is clear that the protocol presented in this experiment enables genetic obtainment and storage for future use in cloning through somatic cell NT when long storage periods are necessary.
It was also demonstrated that despite the problems entailed by longer storage periods, viable cells can be obtained, and a satisfactory blastocyst rate can be achieved with such cells, demonstrating that the protocol is indeed efficient for storage up to 30 days.
Footnotes
Acknowledgments
The authors thank Embrapa 01130600106.02.04, Fundação de Apoio à Pesquisa do Distrito Federal (FAP-DF) 0193.001579/2016, and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) AUX-PE-PNPD 2352/2019 for financial, technological, and structural support.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was funded by Embrapa ID 01130600106.02.04 and Fundação de Apoio a Pesquisa do Distrito Federal (FAP-DF) ID 0193.001579/2016.