
Abstract
There is growing interest in proteomic analyses of tissue biopsies to reveal pathophysiology and identify biomarkers. The current gold standard for collecting tissue biopsies for preserving the proteome and post-translational modifications is flash freezing in liquid nitrogen (LN2). However, in many clinical settings, this is not an option due to unavailability of LN2 nor trained personnel for rapid biospecimen processing. To address this need, we developed a proof-of-concept quick-freeze prototype device to rapidly freeze biospecimens at the point-of-care to preserve the phosphoproteome without the need for LN2. Our objectives were to develop the device, demonstrate the ease of use, confirm the ability to ship through existing cold chain logistics, and evaluate the cooling performance (i.e., cool a tissue sample to <0°C in <60 seconds, below −8°C in <120 seconds, and maintain temperature <0°C for >60 minutes) in the context of preserving the proteome in a tissue biospecimen. To demonstrate feasibility, the performance of the prototype was benchmarked against flash freezing in LN2 using a murine melanoma patient-derived xenograft model subjected to total body irradiation to elicit phosphosignaling in the DNA damage response network. Tumors were harvested and quadrisected, with two parts of the tumor being snap frozen in LN2, and the remaining two parts being rapidly cooled in the prototype quick-freeze biospecimen containers. Phosphoproteins were profiled by liquid chromatography tandem mass spectrometry and quantified by targeted multiple reaction monitoring MS. Overall, the phosphoproteome was equivalent in biospecimens processed using the quick-freeze containers to those using the LN2 gold standard, although the measurements of a subset of phosphopeptides in the device-frozen specimens were more variable than LN2-frozen specimens. The prototype device forms the framework for development of a commercial device that will improve tissue biopsy preservation for measurement of important phosphosignaling molecules.
Introduction
Reliable quantification of phosphoproteins in cancer tissue biospecimens is important for understanding tumor biology and for translation of novel molecularly targeted agents into clinical use. Abnormalities in phosphosignaling networks in cancers are often the (direct or indirect) target of antitumor agents.1–5 As a result, quantifying phosphoprotein activity is used to establish mechanism of action, evaluate pharmacodynamic (PD) responses (e.g., informing dose and scheduling), and monitor compensatory pathways that influence efficacy and toxicity.6–8 Accordingly, the technical capability to quantify phosphorylation networks in biospecimens using mass spectrometry has improved tremendously in recent years.9–12
Phosphosignaling is used extensively by cells to respond to perturbations. Thus, it is not surprising that the phosphoproteome undergoes extensive changes as soon as a tumor is perturbed during biopsy procedures, surgical resection, or chemical fixation.13–17 Previous studies have demonstrated that up to 24% of the phosphoproteome is altered by ischemia, 18 with widespread impacts on phosphotyrosine (pTyr) networks occurring within 5 minutes of resection, altering up to 50% of pTyr sites by more than twofold. 19 Thus, any delay in freezing the tissue biospecimen can be expected to introduce artifacts reflected in molecular data generated using these samples.
Formalin fixation of tumor biospecimens is widely used for tissue preservation, but the process of fixation introduces chemical modifications to the proteome. 20 Nonchemical fixation techniques, such as biomarker and histology preservative chemistry, 21 PAXgene®,22,23 and HOPE® fixation, 24 have successfully measured a limited number of phosphosites at the same levels as fresh frozen tissues, but another comparative study demonstrated worse performance at quantifying phosphorylated human epidermal growth factor receptor 2 than formalin-fixed paraffin-embedded tissue samples. 25 In all fixation techniques, artifacts can occur based on fixation time,13–17 with ischemia continuing as the fixative penetrates the tissue, at the rate of ∼1 mm per hour in the case of formalin. 26
Thus, the current gold standard for collecting tumor tissues for phosphoproteomic analyses involves flash freezing in liquid nitrogen (LN2). Although this approach is readily achievable in some laboratory settings, the majority of clinical sites (e.g., operating rooms, outpatient clinics, or radiology suites) lack the time, expertise, and/or infrastructure to process samples optimally. To effectively preserve the biospecimen in a rapid manner usually requires a dedicated person with the appropriate training and materials. As a result, it is difficult to perform valuable PD studies in clinical trials, as many tumor biospecimens are compromised.
To enable flash freezing of biospecimens in diverse clinical settings, we developed and evaluated a prototype quick-freeze single-use point-of-care biospecimen collection container that can be used with minimal training, minimal time, and no specialized infrastructure. We evaluated the performance of the prototype device (compared with flash freezing in LN2) by comparing phosphopeptide measurements in tumor tissues harvested from a melanoma patient-derived xenograft (PDX) model subjected to total body irradiation to elicit phosphosignaling in the DNA damage response network.
The device offers a potential path toward a low-cost disposable easy-to-use device that enables rapid freezing of biospecimens from clinics (or field applications) to be deposited in biobanks spanning a wide range of resources and technical expertise. Wide adoption and availability of this simple device could improve biospecimen viability and availability for proteomic diagnostics, enable PD studies, and empower oncologists with more precise diagnostic consistency and quality.
Materials and Methods
An expanded materials and methods section is available in the Supplementary Data.
Quick-freeze prototype operation
Twenty copies of the prototype were made for testing using manufacturing methods such as three-dimensional (3D) printing. Dimethyl ether/propane was selected as an aerosol cooling system. The prototype functions by closing the lid and depressing the activation button to release the coolant (Cat. No. SP-133; Freeze Away® Wart Remover, Dr. Scholl's) into a chamber containing the cryovial with the specimen. The pool of coolant keeps the sample frozen while it slowly boils off. Excess fluid and gas are collected in an exhaust system in the lower portion of the device. Performance testing of the prototype was done using a 1 cm3 piece of Spam® (Cat. No. 30074; Hormel) as a tissue surrogate.
PDX sample generation
A Melanoma PDX model was developed and supported by the Preclinical Modeling Core at the Fred Hutchinson Cancer Research Center (FHCRC). An aseptic standardized procedure was followed uniformly: working within a biosafety cabinet, cryopreserved PDX whole tissue was thawed to 37°C, rinsed in Roswell Park Memorial Institute medium, and suspended in Matrigel (Corning). Tissue fragments of 3 × 3 mm were implanted subcutaneously into the right and left flanks of 6- to 8-week-old female immunocompromised non-obese diabetic, severe combined immunodeficiency gamma (NSG™) mice under isoflurane anesthesia.
Pre-emptive analgesia was provided using buprenorphine sustained-release 0.05 mg/kg. The bilaterally implanted mice were subjected to total body irradiation of 6 Gy using a cesium irradiator. One hour post-irradiation, the mice were humanely euthanized, and the tumors, liver, and spleen of each mouse were aseptically collected and quadrisected. Two parts of each tissue were snap frozen in LN2, and the remaining two parts were rapidly frozen in the quick-freeze prototypes and held there for 1 hour before being transferred to an LN2 tank. All work in mice was approved by the institutional animal care and use committee (FHCRC IACUC protocol Roble 50935-200016).
PDX sample preparation
Frozen tumors were cryopulverized using a cryoPREP CP-02 (Covaris), and protein was solubilized in 1 mL of a 6 M urea solution as described, 27 with modifications noted in the Supplementary Data. A pooled reference sample was prepared by combining 200 μg of each individual sample. In total, 500 μg aliquots (global phosphoprofiling and immunomultiple reaction monitoring [immuno-MRM]) or 200 μg aliquots immobilized metal affinity chromatography multiple reaction monitoring (IMAC-MRM) were reduced and alkylated, then incubated at 37°C for 2 hours after LysC addition at 1:50 enzyme–protein ratio by mass and then overnight after trypsin addition at 1:100.
After digestion, samples for targeted MRM analysis were spiked with a mixture of standard isotope-containing synthetic peptides (New England Peptide), as detailed in Supplementary Table S1, and the samples were desalted. Global phosphoprofiling samples were labeled using the TMT10plex™ isobaric label reagent set as described. 27 Individual samples were labeled using channels 1–8 and the pooled reference sample was labeled using channel 9. The independent labeling reactions were pooled together, lyophilized, desalted, and fractionated as described 27 to generate 12 samples, which were dried down and stored at −80°C before phosphopeptide enrichment.
Phosphopeptide and immunoaffinity enrichment
Fractionated samples were enriched for phosphopeptides by immobilized metal affinity chromatography (IMAC) using freshly prepared Fe3+-NTA-agarose beads (Cat. No. 36113; Qiagen) for global phosphoprofiling as described. 27 Separate aliquots of unfractionated samples were IMAC enriched as described 9 for targeted IMAC-MRM analysis. Peptide immunoaffinity enrichment was performed as described,28,29 with modifications noted in the Supplementary Data. Enrichment was performed sequentially using a mixture of 55 antibodies followed by a mixture of 39 antibodies. Antibodies were cross-linked on protein G beads (Cat. No. 28-9513-79; GE Sepharose), and peptide enrichment was performed using 1 μg antibody–protein G magnetic beads for each target.
In total, 500 μg of trypsin-digested lysate resuspended in 200 μL phosphate buffered saline (PBS) +0.03% 3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate (CHAPS) (pH was adjusted to 8.0 with 5 μL of 1 M Tris) was inputted to each enrichment. Beads were mixed in the incubation plate, washed twice in PBS buffer +0.03% CHAPS, washed once in 1/10 × PBS, and peptides were eluted in 26 μL of 5% acetic acid/3% MeCN/50 mM citrate. For sequential enrichment, once the beads were removed from the initial overnight incubation plate (by the KingFisher), the next set of beads and antibodies was immediately added (to the incubation plate), and the plate was placed on a tumbler at 4°C for overnight incubation.
Global phosphoprofiling by nanoliquid chromatography-tandem mass spectrometry
Labeled and fractionated global phosphoprofiling samples were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) on an Easy-nLC 1000 (Thermo Scientific) coupled to an Orbitrap Fusion Tribrid mass spectrometer (Thermo Scientific) operated in positive ion mode as previously described 27 using parameters found in the Supplementary Data. Raw MS/MS spectra from the analysis were searched against reviewed Human Universal Protein Resource (UniProt) sequence database release 2019_06 using MaxQuant/Andromeda. 30 Data normalization was performed by scaling the TMT channels of the individual samples (channels 1–8) to the pooled reference channel (channel 9), and then these ratios were log(2) transformed followed by batch effect correction using ComBat 31 in R.
Targeted LC-MRM-MS analysis by nano-LC-MS/MS
Targeted LC-MRM-MS analysis was performed as described9,28,32 by a trap-elute configuration on a nanoLC-2D and cHiPLC-Nanoflex system (Eksigent Technologies, Dublin, CA) coupled to a 5500 QTRAP mass spectrometer (SCIEX, Foster City, CA) by an Advance CaptiveSpray source (Michrom Bioresources, Auburn, CA). MRM peak integration was performed using Skyline, 33 with the sum of all transitions used for quantification. The integrations were manually inspected to ensure correct peak detection, absence of interferences, and accurate integration.
Public access to data
Raw data from the global phosphoprofiling experiments have been deposited to the ProteomeXchange Consortium 34 through the PRIDE 35 partner repository with the data set identifier PXD029844. All targeted MRM data have been deposited in Panorama Public 36 at https://panoramaweb.org/PO4_PDX_MRM.url.
Results
Development of a quick-freeze biospecimen container prototype
Development of the quick-freeze biospecimen collection device consisted of four phases: (1) theoretical design, (2) prototype development, (3) performance testing, and (4) proof-of-concept testing in the end-use application setting. Based on existing knowledge of phosphoprotein lability 18 and considering practical limitations of biospecimen collection, we established the following performance goals for a useful device: (1) cool a sample to <0°C in 60 seconds from receipt, (2) cool to less than −8°C in 120 seconds, and (3) maintain temperature <0°C for at least 1 hour (for a tissue sample size ∼1 cm3). We expect that by meeting these goals, the phosphoproteome will remain intact and the samples will remain frozen long enough to enter a cold chain logistics pathway.
The theoretical design was constructed by incorporating numerous “user” (i.e., surgeons, pathologists, laboratory technicians, research coordinators, biorepository coordinators, and pharma) inputs and considerations. Several coolant methods were considered for both performance and cost target requirements, and initial testing found that an aerosol cooling system (using dimethyl ether/propane or tetrafluoroethane) was more effective than an endothermic chemical solution (using calcium ammonium nitrate) at quickly cooling a tissue specimen. As a proof of concept, dimethyl ether/propane was selected due to having a low boiling point (−32°C) while also being able to form a liquid pool at atmospheric pressure.
At the conclusion of the theoretical design phase, the final device architecture and user interface were chosen based on the analysis of nonfunctional prototype models, and a prototype was built from the final design (Fig. 1). Twenty copies of the prototype were made for testing using conventional and 3D printing manufacturing methods (Supplementary Fig. S1).

Schematics of a prototype that can rapidly freeze tissue specimens and keep them frozen. The prototype is 10 × 10 × 16.5 cm with a chamber for a 1.0 mL cryovial, a locking lid, and an activation button that releases the coolant into the cooling chamber containing the cryovial and tissue sample. The prototype functions by closing the lid and depressing the single-use activation button to release the coolant from a pressurized container into a chamber containing the cryovial with the specimen. The released liquid rapidly cools its surroundings until it can form a pool (1–2 seconds after release), and the expansion and evaporation of the pool cools the outside of the vial, which in turn freezes the tissue inside the vial. The pool of coolant keeps the sample frozen while it slowly boils off. Excess fluid and gas are collected in an exhaust system in the lower portion of the device.
The performance of the prototype quick-freeze biospecimen container was tested using three independent 1 cm3 aliquots of commercially available processed meat (Spam®) as tissue surrogate samples. The samples were placed into the prototype and the core temperature was monitored using a temperature probe. Results show that on average, the core sample temperature decreased to 0°C within 69.2 seconds, to −8°C within 150.2 seconds, and remained <0°C for 72.3 minutes (Fig. 2), exceeding one of the goals and approaching the others. Upon showing acceptable performance, we next evaluated the device for preservation of the phosphoproteome using PDX tumor tissue biospecimens harvested from PDX-bearing mice.
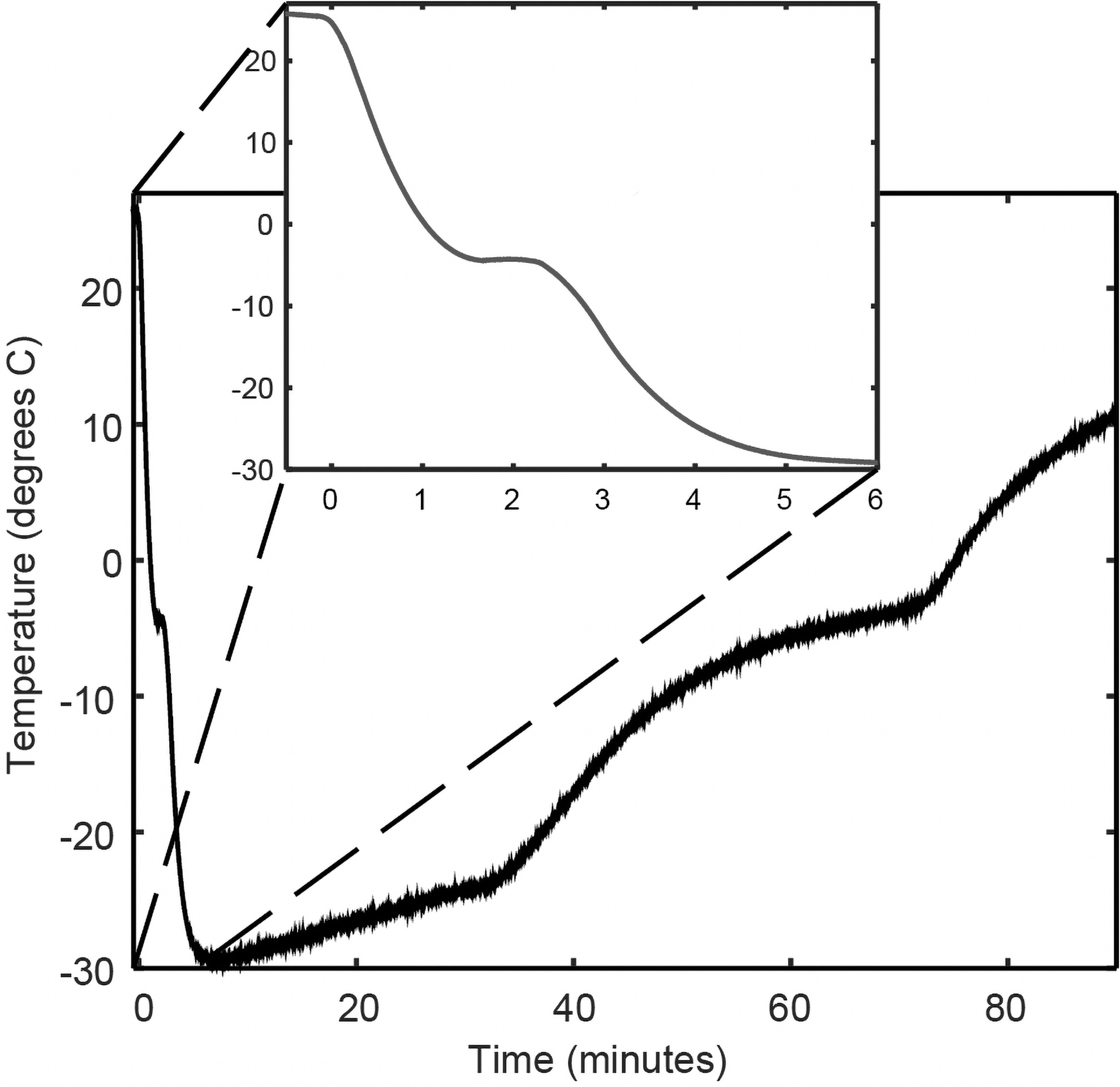
Performance testing of the quick-freeze prototype using a 1 cm3 surrogate tissue sample. The internal temperature of the tissue surrogate sample was recorded for 90 minutes in three independent trials. The inset focuses on the temperature from 0 to 400 seconds, where on average, the internal temperature of the samples fell <0°C within 70 seconds and below −8°C within 150 seconds. The larger graph shows the test sample temperature as a function of time from 0 to 90 minutes, showing that on average, the sample remained <0°C for >70 minutes.
The sample preservation device preserves the phosphoproteome during sample procurement
To demonstrate the utility of the device for preserving the phosphoproteome, we conducted a proof-of-concept study (Fig. 3) using mass spectrometry to quantify phosphorylated peptides in biospecimens collected in the prototype device compared with those collected using the gold standard flash freezing in LN2. We chose to use a human-in-mouse melanoma PDX model to minimize biological noise and study the technical performance of the device. The melanoma PDX model shows minimal heterogeneity (within and between tumors) in the relative contributions of different cell types (e.g., mouse stroma content, tumor cellularity, necrosis, vascularity; Supplementary Fig. S2), minimizing biological variation that could affect the number or expression levels of phosphorylated peptides.
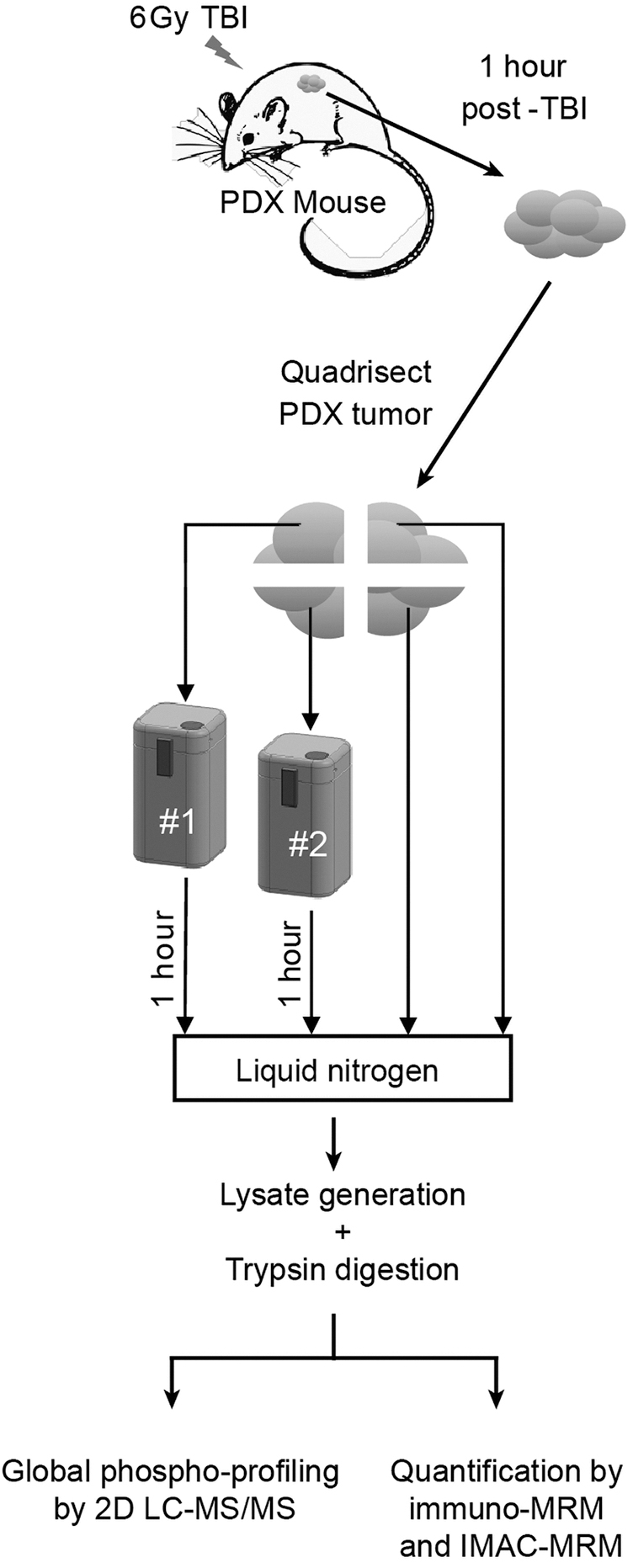
Workflow for comparing phosphoproteome integrity in device-frozen PDX tumor samples compared against snap freezing. Seven PDX mice bearing melanoma tumors were subjected to total body irradiation to activate cellular phosphosignaling in response to DNA damage. One hour postirradiation, PDX tumors were harvested and quadrisected. Two parts of the tumor were snap frozen in LN2, and the remaining two parts were rapidly frozen in the quick-freeze prototypes for 1 hour, providing a replicate for both approaches in each mouse and helping to account for tumor microheterogeneity. In total, 10 PDX tumors were harvested for profiling by LC-MS/MS phosphoprofiling as well as targeted MRM MS-based quantification of a panel of phosphosites that respond to DNA damage. Phosphoprofiling was performed by isobarically labeling the individual samples, mixing them together, and then fractionating the mix before LC-MS/MS analysis. Targeted phosphosite quantification was performed by spiking in heavy isotope-labeled standards to the target phosphosites, which were then enriched by IMAC or antipeptide mAbs before LC-MRM-MS analysis. IMAC, immobilized metal affinity chromatography; LC-MS/MS, liquid chromatography-tandem mass spectrometry; LN2, liquid nitrogen; MRM, multiple reaction monitoring; PDX, patient-derived xenograft.
Tumors were harvested from the melanoma PDX model after total body irradiation and quadrisected for freezing in the prototype device or in LN2 (details for each specimen, including the host mouse, irradiation conditions, and cold ischemia time, are in Supplementary Table S2). We then assessed the equivalence of the phosphoproteomes in biospecimens processed using the quick-freeze containers versus the LN2 gold standard. Global LC-MS/MS phosphoprofiling was performed using an isobaric-labeling (TMT) approach 37 to analyze 40 samples from the 10 PDX tumors relative to a reference sample comprising a pool of an equal amount (by mass) of each individual sample in the study.
The samples were fractionated, and phosphopeptides were enriched by iron metal affinity chromatography (IMAC) before analysis by LC-MS/MS (Fig. 3). In addition to global phosphoprofiling, we used targeted multiplexed MRM mass spectrometry-based assay panels for precise relative quantification of specific phosphopeptides by enriching for phosphopeptides by IMAC (IMAC-MRM) 9 or using antipeptide antibodies (immuno-MRM).28,32
Global analysis quantified a total of 11,346 phosphosites in at least one PDX tumor sample (Supplementary Fig. S3). To combine results from all samples, TMT reporter ion intensities of the individual samples (Supplementary Table S3) were normalized by the common reference sample and then batch corrected using established approaches, 31 yielding 3596 phosphosites quantified in all of the samples (Supplementary Table S4). In the targeted MRM approach, 40 phosphopeptides were quantified above the lower limit of quantification by IMAC-MRM and 19 by immuno-MRM (Supplementary Table S5) in at least one of the pieces in all tumor samples.
Phosphoproteome profiles were used to assess the equivalence of biospecimens processed using the quick-freeze containers versus the LN2 gold standard. Phosphopeptide measurements were comparable between device-frozen samples compared with LN2-frozen samples, as measured by global phosphoprofiling, IMAC-MRM, and immuno-MRM (p-values equal to 0.08, 0.97, and 0.94, respectively; Fig. 4). Furthermore, hierarchical clustering and principal component analysis of the phosphoprofiling results showed no predominant clusters or systemic difference based on freezing approach (Supplementary Fig. S4).
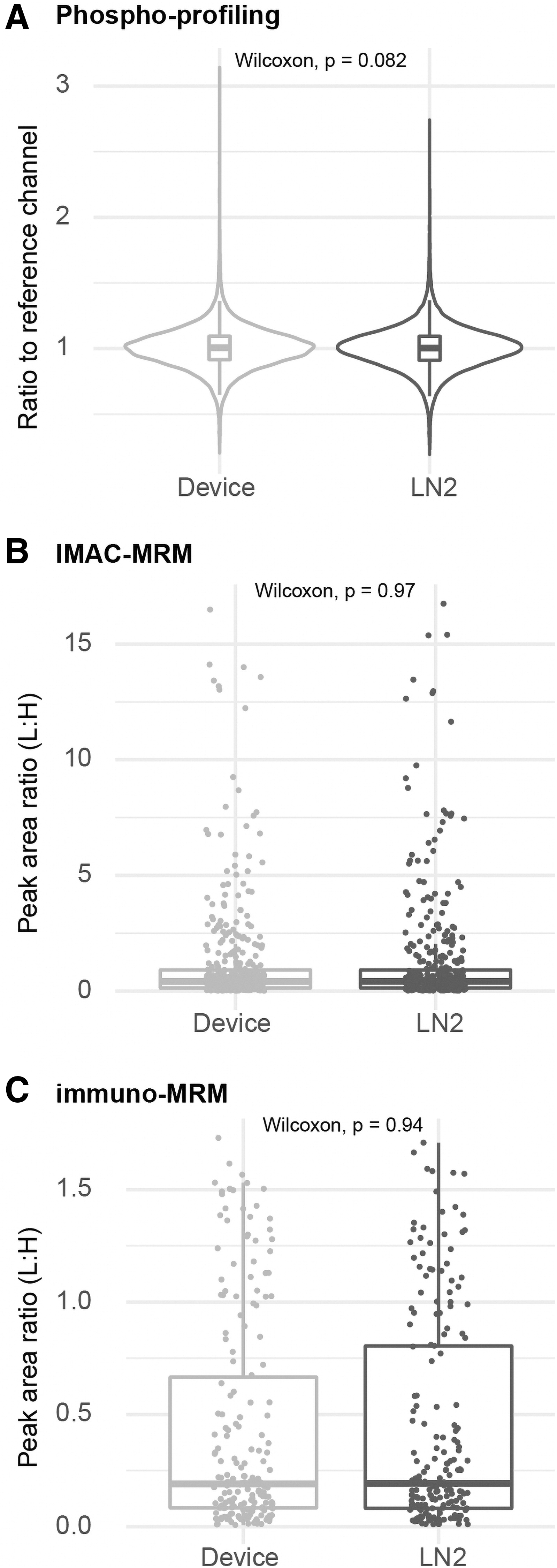
Phosphosite quantification is not affected by using the prototype device for preserving specimens.
We found the majority of individual phosphosites measurements in samples frozen by the device were within 20% of those frozen by LN2, as measured by phosphoprofiling (median difference of 1% ± 16%), IMAC-MRM (3% ± 15%), and immuno-MRM experiments (−2% ± 21%; Fig. 5). A small percentage of individual phosphosites was found to be significantly different between the freezing techniques (false discovery rate <0.05) (n = 307 [8.6%]; Supplementary Table S4), IMAC-MRM, and immuno-MRM (n = 7 [17.5%] and n = 1 [6.2%]; Supplementary Table S5); no phosphosites were significantly different in more than one experiment.

Difference of phosphosite levels between LN2 and device-frozen PDX tumor tissues. Histograms showing the distribution of the pairwise comparisons (% difference) for each phosphosite measured in samples preserved by the prototype device and LN2 for
In addition to evaluating the differences in the phosphosite measurements in specimens frozen by the two approaches, we also evaluated the variability of the phosphosite measurements by measuring the within-tumor variability (using the percentage difference across the two sections of tumor measured by each method), and between-tumor variability (using the percentage difference across the 10 tumors). Tightly controlled ischemia time across the 10 tumors (between 5 and 11 minutes; Supplementary Table S2) allowed for between-tumor comparisons.
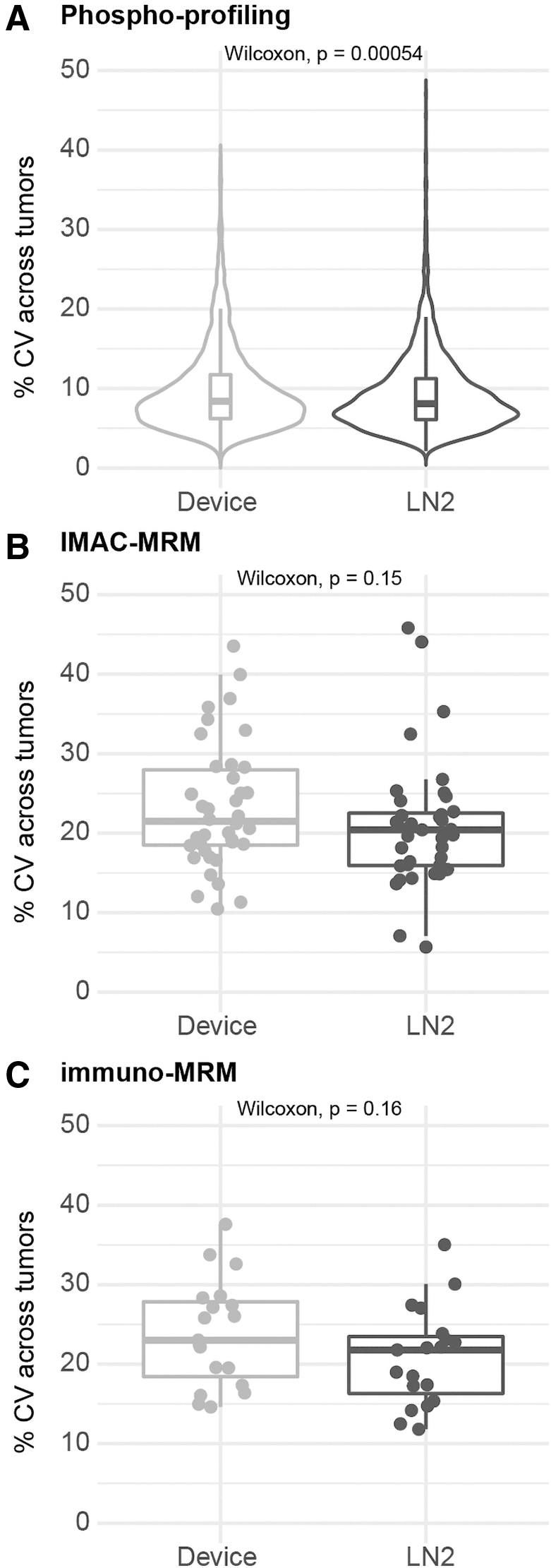
Across the phosphoproteome, the within-tumor variability was slightly higher in the device-frozen tumor pieces by phosphoprofiling (median variation of 11.1% vs. 8.3%, p < 2.2 e-16), IMAC-MRM (median variation of 12.3% vs. 9.9%, p = 0.0033), and immuno-MRM (median variation of 16.6% vs. 12.8%, p = 0.0023; Fig. 6). The between-tumor variability was also slightly higher in the individual device-frozen tumors by phosphoprofiling (median variation of 8.4% vs. 8.1%, p = 0.0005), IMAC-MRM (median variation of 22.0% vs. 20.4%, p = 0.15), and immuno-MRM (median variation of 23.0% vs. 21.8%, p = 0.16; Fig. 7).
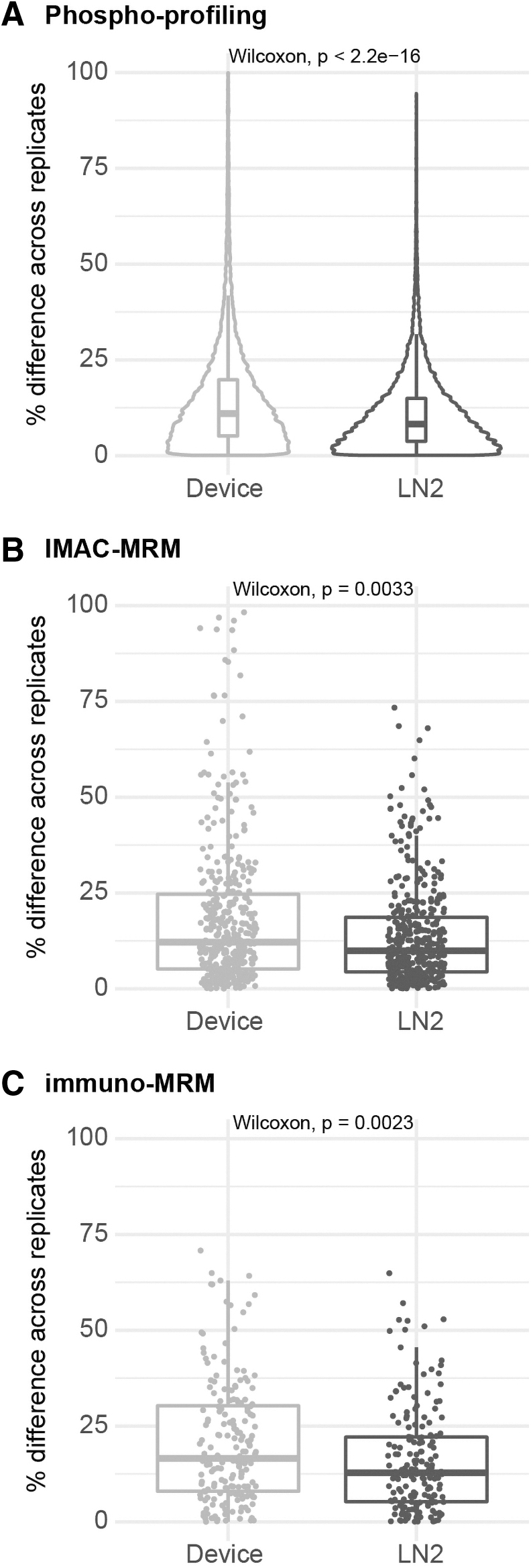
Variability of phosphosite measurements within tumor in device-frozen PDX tumor tissue pieces compared with snap-frozen pieces. Variability in phosphosite quantification reported as the percentage difference in the replicate tumor pieces measured by
Variability of phosphosite measurements between tumors in device-frozen PDX tumor tissue pieces compared with snap-frozen pieces. Variability in phosphosite quantification reported as the percentage difference across 10 individual tumors measured by
The variability of the individual phosphosites between tumors correlated well across the two freezing methods (R 2 = 0.65, 0.66, and 0.68, respectively; Fig. 8), meaning that those phosphosites with large variability in the device-frozen samples also had large variability in the LN2-frozen samples. It was observed that some of the devices did not completely dispense all the cooling liquid during the process of freezing the PDX tumor (Supplementary Table S2), which may have compromised the performance of the device during freezing and led to higher variability in the device (Supplementary Fig. S5).
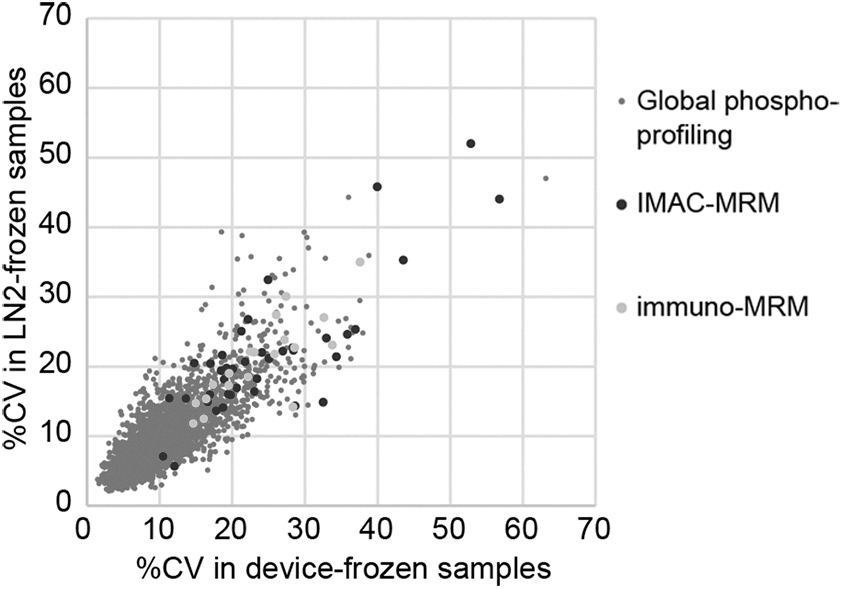
Variability of individual phosphosite measurements across tumors in device-frozen PDX tumor tissue pieces compared with snap-frozen pieces. The variability in phosphosite quantification reported as the percentage CV across individual tumors in the global phosphoprofiling, IMAC-MRM, and immuno-MRM measurements. Percentage CV was calculated as the standard deviation of the measurements across the 10 individual tumor samples (averaged between the two replicate tumor pieces) divided by the average of the measurements.
To examine whether the preanalytical effects are specific to phosphorylation, we compared unmodified peptide measurements (in the device-frozen and LN2-frozen samples) that were fortuitously measured in the phosphoprofiling experiment (n = 528; Supplementary Table S4) and targeted in the immuno-MRM assay panel (n = 36; Supplementary Table S5).
Compared with phosphopeptide measurements, unmodified peptide measurements showed less difference between the freezing techniques (median difference of 0% ± 14% for global profiling, and 0% ± 12% for immuno-MRM), and the variation in the measurements across the two replicates was not significant (median variation of 8.2% vs. 8.4%, p = 0.54 and 11.1% vs. 10.5%, p = 0.16; Supplementary Fig. S6). These results underscore the greater sensitivity of the phosphoproteome (vs. unmodified proteins) to preanalytical sample handling in tissue specimen collection.
Discussion
In this project, we developed a prototype device for freezing tissue biospecimens to minimize preanalytical variation affecting phosphoproteomic analyses. This is a first step toward development of a commercial product capable of filling a gap in the cold chain in clinical settings such as operating rooms, outpatient clinics, radiology suites, or in the field, where LN2 is typically unavailable to preserve biological samples. The device has a simple user interface, and it rapidly freezes tissue samples and keeps them cold in a self-contained single-use container until they can be transported to cryostorage.
We showed feasibility in preserving the majority of the phosphoproteome using the quick-freeze prototype with comparison with the gold standard snap freezing using LN2. Higher variability in the device-frozen tumors was likely the result of inconsistent release of coolant, an issue that can be resolved in future versions of the product.
Improvements to the prototype to enable a commercial-quality unit include reducing cost, improving performance (e.g., more consistent release of coolant), reducing the footprint, using a nonflammable gas, and incorporating the ability to recharge units with new coolant canisters. The addition of a data logger could capture the temperature profile near the cryovial to ensure the sample was properly cooled. We expect a more consistent release of the coolant across multiple devices as manufacturing goes from prototypes to production, including observing tighter tolerances in manufacturing and custom designing a coolant canister for optimum performance.
The next generation version of the device would be made of molded expanded polystyrene, and the plastic components would be primarily blow molded to minimize the amount of plastic used. Using these methods, we will be able to create a product with a low cost of manufacturing. Most of the cost will be from the coolant canister itself, which we can optimize for our application.
Future study will be needed to validate the commercial device and incorporate biospecimens into existing workflows while also addressing possible biobanking concerns, such as standardized collection protocols, broad informed consent, and storage requirements. Finally, further testing will be required in the clinic or procedure room to evaluate the performance of the device in a real-use setting.
Conclusion
In summary, we developed a prototype device and demonstrated proof of principle in filling the gap in the cold chain for biological tissue specimens. The device has a simple user interface, and it rapidly freezes tissue samples and keeps them cold in a self-contained single use container. We demonstrated the feasibility of the device by benchmarking the performance against the gold standard of flash freezing in LN2, showing that the majority of the phosphoproteome could be stabilized using the quick-freeze device. Future versions of the prototype can be improved to improve performance and overcome the obstacles to commercial deployment.
Footnotes
Authors' Contributions
J.J.K. and J.R.W. contributed to investigation (equal), data curation (equal), formal analysis (equal), and writing—original draft (equal). A.W., G.C., E.H., G.M., G.L., B.S., and T.D.L. were involved in conceptualization (equal) and methodology (equal). ![]() ., C.W.L., E.A.C., and L.Z. carried out investigation (equal), data curation (equal), and formal analysis (equal). S.T. was involved in supervision (equal), conceptualization (equal), and methodology (equal). A.G.P. was involved in supervision (equal), conceptualization (equal), methodology (equal), and writing—original draft (equal).
., C.W.L., E.A.C., and L.Z. carried out investigation (equal), data curation (equal), and formal analysis (equal). S.T. was involved in supervision (equal), conceptualization (equal), and methodology (equal). A.G.P. was involved in supervision (equal), conceptualization (equal), methodology (equal), and writing—original draft (equal).
Author Disclosure Statement
A.W., G.C., E.H., G.M., G.L., B.S., and S.T. are employees of Product Creation Studio. No competing financial interests exist for the remaining authors.
Funding Information
This study was supported in part by the National Cancer Institute (NCI) of the National Institutes of Health, Innovative Molecular Analysis Technologies Program (R21CA225507-02), the Preclinical Modeling Core Shared Resource of the Fred Hutch/University of Washington Cancer Consortium (P30 CA015704), the NCI Academic-Industrial Partnerships Program (R01CA235575), and the NCI Clinical Proteomics Tumor Analysis Consortium Initiative (U24CA160034).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.