
Abstract
Oocyte vitrification has become a widely adopted method in clinical practice. However, the solidification behavior and its impact on oocytes during the ultrarapid cooling process remain poorly understood. In this study, we established a system and methodology to observe crystallization behavior in oocytes during quench cooling and warming. Subsequently, the threshold concentration of cryoprotective agents (CPAs) required for oocyte vitrification was determined through a visualization method. The results demonstrated that the ice front could not be observed in the image sequence when using 16.5% DMSO +16.5% EG during high-speed quench cooling (2821.58°C/min). Finally, oocytes were encapsulated with an antifreezing hydrogel (7.5% EG +7.5% DMSO +0.5% alginate) and subjected to high-speed quench cooling. No ice crystals appeared in the antifreezing hydrogel-encapsulated oocytes at a low concentration of osmotic CPA (2.4 M). This research opens up new possibilities for oocyte vitrification with a reduced concentration of CPA.
Introduction
Oocyte cryopreservation is a vital component of assisted reproductive technology, 1 serving to preserve fertility for women undergoing cancer treatment or facing other factors that may delay childbearing.2,3 In recent years, it has found extensive application in egg donation programs and offers a solution for managing surplus oocytes in assisted reproduction treatments. 4
Furthermore, it plays a crucial role in the conservation of endangered species and valuable breeding resources. 5 In clinical settings, stage MII oocytes, characterized by maturity and fertility, are the standard for cryopreservation. 6 Currently, the commonly used oocyte cryopreservation methods include slow freezing and vitrification.7,8 Vitrification is preferred because it prevents intracellular and extracellular ice crystal formation, which improves the survival and developmental potential of oocytes.9,10
Vitrification typically involves rapidly cooling samples with high concentrations of cryoprotective agents (CPAs) by directly plunging them into liquid nitrogen, achieving ultrarapid cooling. However, the solidification behavior and its impact on cells during vitrification or partial vitrification remain unclear. Currently, the observation of freezing processes using microscopy is constrained by the design of the cryogenic stage, and most studies are conducted at relatively low cooling rates.
Ninagawa et al 11 utilized a cryomicroscopy system to investigate strawberry geranium cells at a slow (1–100°C/min) cooling rate, revealing certain characteristics of intracellular ice growth. While rapid cooling visualization relies on high-speed camera systems. Stott et al 12 captured the intracellular ice formation (IIF) of bovine pulmonary artery endothelial cells through cryomicroscopy during rapid freezing, recording at a frame rate of up to 16,000 fps. Notably, the vitrification process of oocytes under microscopy observation has not been reported.
The use of high concentrations of CPAs during vitrification is necessary to prevent the formation of ice crystals. However, in practical applications, the CPA concentration often exceeds the threshold required for vitrification, leading to unnecessary toxicity and osmotic damage to cells. 13 Liu et al 14 reduced the CPA concentration from 15%EG +15%DMSO to 7.5%EG +7.5%DMSO for cryopreservation. The survival rate and developmental potential of oocytes after warming were found to be not significantly different from those preserved with the higher concentration of 15% EG +15% DMSO. Despite these advancements, there remains a lack of experimental methods to accurately determine the threshold concentration of CPAs required for vitrification in preservation processes.
High concentrations of CPA often lead to severe damage to cells, and finding ways to reduce CPA concentration during cryopreservation has been a significant challenge for scientific researchers. In recent years, hydrogels have gained widespread use in cryopreservation due to their excellent biocompatibility and three-dimensional network structure that impedes the growth of ice crystals. Huang et al 15 utilized alginate hydrogels to encapsulate pluripotent stem cells for vitrification preservation, demonstrating that gene and protein expression of cell differentiation indicated the retention of functional properties in the preserved stem cells. The microencapsulation of alginate hydrogels proved effective in protecting cells from cryogenic damage.
In another study, Shi et al 16 prepared supramolecular polyvinyl alcohol (PVA)-glycerol hydrogels by mixing PVA with water and glycerol. In addition, Chen et al 17 utilized ethylene glycol (EG) to replace water in alginate hydrogels, enhancing the antifreezing properties of the hydrogels. Building on these insights, a combination of the two methods, namely “physical mixing” and “solvent replacement,” holds the potential to create a hydrogel with antifreeze capabilities. Presently, there are no reported instances of cryopreserving hydrogel-encapsulated oocytes, nor is there visualization of hydrogel-encapsulated oocytes during high-speed quench cooling and warming.
This study employs a high-speed quench cooling device to observe ice crystals' behavior in oocytes during quench cooling and warming. First, we established a high-speed quench platform and the visualization method. Then, the crystallization behavior of oocytes on the high-speed quench cooling device was observed to determine the threshold concentration of CPA required for oocytes vitrification. Finally, oocytes are encapsulated with an antifreezing hydrogel, and the ice suppression mechanism of the hydrogel is analyzed.
Materials and Methods
Chemicals
Pregnant mare serum gonadotropin (PMSG) and human chorionic gonadotrophin (hCG) were purchased from Ningbo Hormone Products. Medium 199, M2 Medium, alginate, sodium citrate, and HEPES were purchased from Gibco.
Oocyte collection
All experimental procedures received approval from the Animal Research Institute Ethics Committee of Shanghai Jiao Tong University School of Medicine. Six-to eight-week-old female ICR mice underwent stimulation with PMSG and hCG to induce significant follicular maturation and ovulation.
The cumulus cells were removed by hyaluronidase (1 mg/mL), and the cumulus oocyte complexes were placed in hyaluronidase at 37°C for 1–2 minutes, and then gently blown with a pipette until the granulosa cells were completely denuded, 18 then thoroughly washed in droplets of M2 culture solution, repeated three to five times, to ensure complete removal of hyaluronidase. This process was observed under a stereomicroscope, and finally oocytes with intact morphology, homogeneous cytoplasm, and smooth surface of polar bodies were selected and placed into microdrops of M2 culture medium (M2 Medium +100 IU/mL Penicillin–Streptomycin). The collected oocytes were used for subsequent experiments and the flow chart is shown in Figure 1.
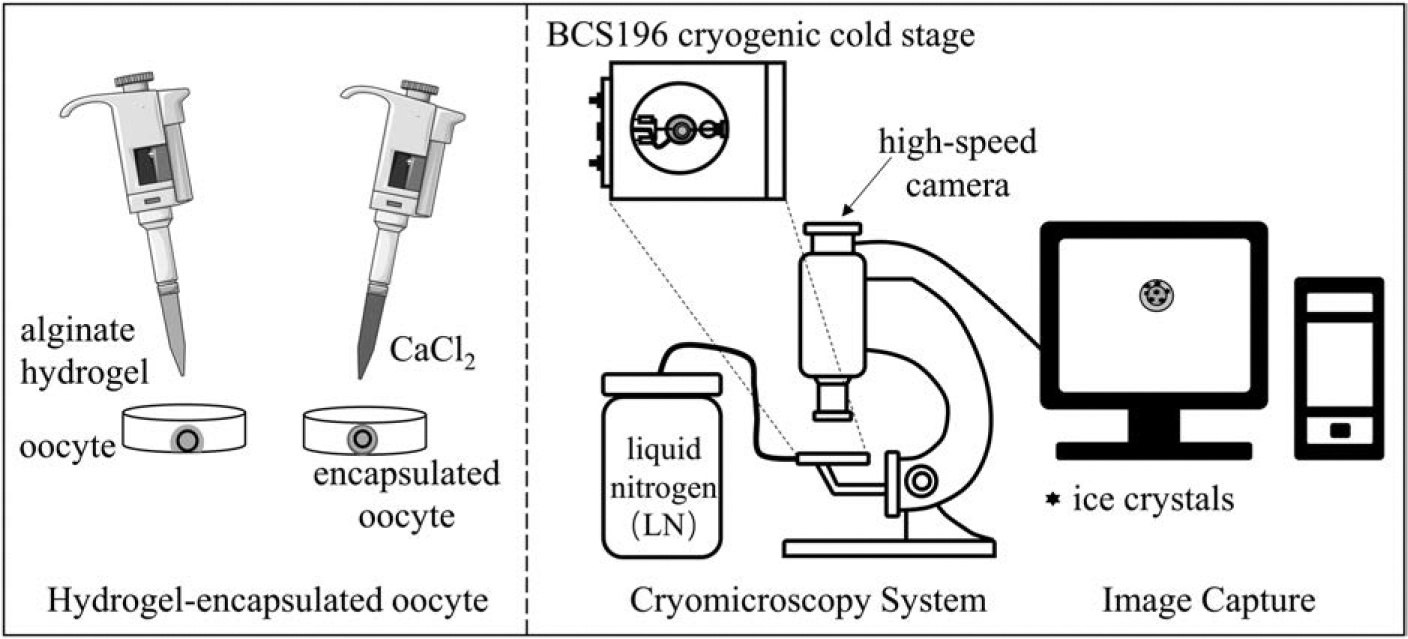
The flow chart of the experiment.
Cryomicroscopy system
The cryomicroscopy system utilized a BX51 TRF microscope (Olympus, JP) equipped with a BCS196 cryogenic cold stage, LNP95 automatic cooling system, T95 temperature control system (Linkam Scientific Instruments), and a high-speed camera (OSG030-815UC, YVSION). The high-speed camera had effective pixels of 300,000 (640 × 480 pixels), a frame rate of 815 fps, an exposure time range of 16 μs to 1 second, and a signal-to-noise ratio of 38 dB. The camera was connected to the computer's USB3.0 interface through a data transmission cable. For high-speed, real-time, uncompressed complete video recording, an M.2 NVMe solid-state drive with a read/write speed of 3000–4500MB/s and a capacity of 1TB was employed.
High-speed quench cooling methods
High-speed quench cooling was achieved using the BCS196 cryogenic cold stage, which primarily consists of a quick push handle, sample holder, sapphire glass, copper block, and silver block, as depicted in Figure 2. A piece of sapphire glass with a diameter of 7 mm and a thickness of 0.3 mm was embedded and securely fixed in the ring of the sample holder.

High-speed quench and cool device. 1. quick push handle 2. copper block 3. sample holder 4. sapphire glass 5. silver block.
As the high-speed quench cooling process is uncontrolled, it was imperative to measure the actual cooling rate. To accomplish this, a 25 μm diameter T-type thermocouple wire was affixed to the sample holder, and 1 μL of the solution was dropped onto the center of the sapphire glass, with the temperature probe intruding into the liquid. The data acquisition instrument (Agilent 34970A) was connected, and the acquisition frequency was set to 50 ms. The sample was then driven by the quick push handle, placed on the silver block precooled to −180°C until the temperature of the thermocouple stabilized, and subsequently, the sapphire glass was pulled back to the copper block.
Before initiating the experiment, the silver block (4) was cooled to −180°C using liquid nitrogen, while the sample holder (2) remained at room temperature. During the experiment, 1 μL of BS (20% fetal bovine serum + culture medium 199) was deposited onto the sapphire glass (3). Subsequently, 5 oocytes were transferred in minimal volume into the droplet and covered with an 8 mm diameter coverslip. Initially, the sapphire glass containing the oocytes was positioned on the sample holder (2) at room temperature.
Once all preparations were complete, the sapphire glass (3) on the sample holder (2) was rapidly pushed onto the precooled silver block (4) using the quick push handle (1) while activating the high-speed camera for video recording. Focus adjustments were made, and the system was held for 2 minutes. Finally, the sample was heated to 20°C at a rate of 150°C/min, then ending the recording. 19 The entire system relied on cooling with liquid nitrogen and rewarming with electric heating.
Determination of critical CPA concentration for vitrification
To determine the critical concentration of CPA required for achieving vitrification in oocytes, five different vitrification solutions (VS) were employed for high-speed quench cooling, as outlined in Table 1. Morphologically intact oocytes were selected, and each was transferred to 300 μL of equilibrium solution (ES, 7.5% DMSO +7.5% EG + BS) for 6 minutes. Subsequently, they were transferred to 300 μL of VS1–VS5 for an additional 1 minute. Rapid transfer to 1 μL droplets of each respective VS1–VS5 on sapphire glass followed, and coverslips were applied to complete the CPA loading process.
The Composition of Vitrification Solution
BS, base solution; EG, ethylene glycol; VS, vitrification solution.
The quench cooling of the five groups of oocytes was then performed as described in section High-speed quench cooling methods, with each experiment repeated three times for each group. Image sequences were captured from the high-speed camera, and the solution concentration corresponding to the point where the movement of the ice front could no longer be observed was determined as the critical CPA concentration for vitrification.
Vitrification of alginate hydrogel-encapsulated oocytes
Base solution (BS): 20% fetal bovine serum (FBS) + Medium 199.
Sodium alginate solution: 100 mg of sodium alginate powder was weighed on an electronic scale into a 50 mL centrifuge tube, added to BS, mixed well, and fixed to 20 mL to prepare a 0.5% concentration of sodium alginate solution.
CaCl2 solution: 333 mg of anhydrous calcium chloride powder was weighed on an electronic scale into a 50 mL centrifuge tube, added to BS, mixed well, and allowed to volume to 20 mL to formulate a final 0.15 M CaCl2 solution.
To prepare alginate hydrogel-encapsulated oocytes, 1 μL of 0.5% sodium alginate solution was dispensed onto a sapphire glass, and a single intact oocyte was carefully selected and transferred to the sodium alginate droplet in minimal volume. Subsequently, 50 μL of 0.15 M CaCl2 solution was added above the droplet, and the crosslinking process was allowed to proceed for 3 minutes.
For the antifreezing alginate hydrogel, the composition included 0.5% sodium alginate +7.5% DMSO +7.5% EG + BS. An intact oocyte underwent prehydration in a 1 M trehalose solution for 1 minute, followed by hydrogel encapsulation using the same method as described above. After crosslinking was completed, 50 μL of antifreezing alginate solution was added dropwise outside of the gel for solvent replacement.
The alginate hydrogel-encapsulated and antifreezing alginate hydrogel-encapsulated oocytes were positioned on the sapphire glass, and the subsequent cooling and warming procedures were carried out as described in section High-speed quench cooling methods.
Image acquisition and processing
The freezing and warming process was observed using a 10 × objective lens. The camera starts shooting at the same time as the cooling process starts. The high-speed camera takes 815 pictures per second, and the exposure time is set to 1 ms. At the end of the experiment, the recorded high-speed video was exported frame-by-frame using Kinovea software. 20
Results
The cooling rate of the high-speed quench cooling
The cooling and warming curves for the high-speed quench cooling device are shown in Figure 3. The average cooling rate of the high-speed quench cooling device during the cooling process reached 2821.58°C/min, reaching a maximum instantaneous rate of 5620.80°C/min.
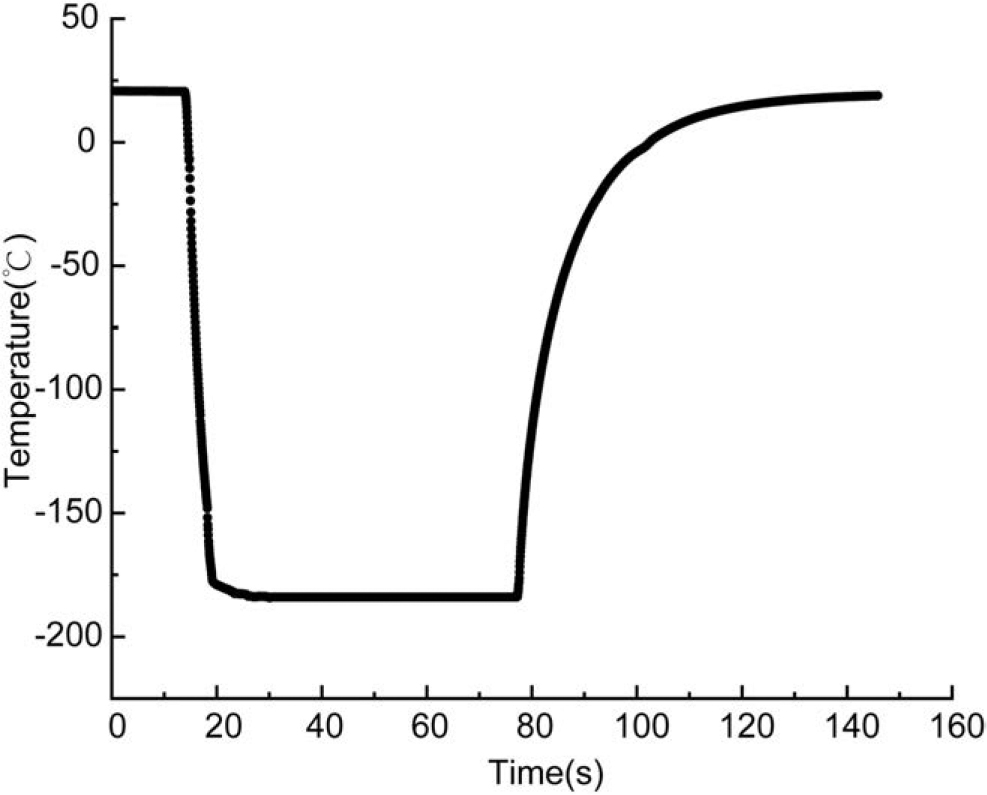
Temperature curve during high-speed quench cooling and warming.
Visualization of high-speed quench cooling and warming of oocytes
The oocytes underwent freeze-thaw cycles utilizing the optimized high-speed quench cooling stage, and the images of the oocytes during the cooling and warming processes are depicted in Figure 4. The formation and melting of ice crystals both inside and outside the oocyte are clearly observed throughout the freeze-thaw cycles. In Figure 4a, intracellular ice emerged from the oocytes during the cooling process upon encountering extracellular ice.

Images of oocytes cooling and rewarming processes.
Upon warming, the sample developed cracks at −180°C due to thermal stress.21,22 We have confirmed that the optimized cryomicroscopy system not only achieves ultrahigh cooling rates but also captures the interaction between oocytes and ice crystals during both cooling and warming. This provides a foundation for determining the critical concentration of CPAs in subsequent experiments.
Critical CPA concentration for vitrification
The oocytes were loaded with five CPAs and subsequently subjected to high-speed quench cooling, and the images of the cooling process are presented in Figure 5. In the VS1 group, point-shaped ice crystals randomly emerged during the cooling process, growing and fusing to occupy the entire field by 6 seconds. The field exhibited varying shades, with black spots scattered, each having an initial nucleus as the center, and relatively bright black ice crystals, indicating local vitrification. Cracks appeared at 20.1 seconds due to thermal stress.

Intracellular and extracellular ice crystals of oocytes during high-speed quench cooling in different VS groups. Scale bar: 50 μm. VS, vitrification solution.
The VS2 group exhibited a similar ice crystal growth pattern to the VS1 group. However, an increase in solute concentration led to a higher number of punctate ice crystals with decreased diameter and an overall lighter color. In the VS3 group, punctate ice crystals were generated during the cooling process, with numerous smaller diameter ice crystals appearing in the field at 5.8 seconds. The elevated solution concentration limited the continued growth of the punctate ice crystals, and at 7.3 seconds, the granular ice crystals fused, rendering the entire field smooth and transparent.
VS4 exhibited only a few punctate ice crystals with a smaller diameter compared with the VS1–VS3 groups. The high solute concentration inhibited ice crystal growth but did not entirely prevent it. Subsequent ice crystal growth led to an increase in the concentration of surrounding solution, allowing the unfrozen solution to vitrify. In the VS5 group (16.5% DMSO +16.5% EG), complete vitrification was achieved, with no ice crystal growth observed in both the internal and external oocytes.
High-speed quench cooling of hydrogel-encapsulated oocytes
The high-speed quench cooling processes of oocytes encapsulated with alginate hydrogel and antifreezing alginate hydrogel are illustrated in Figure 6. Figure 6a depicts the formation and propagation of ice crystals in 0.5% alginate hydrogel-encapsulated oocytes during quench cooling. Ice crystals entered from the right side of the field at 0.9 ms, propagating within the hydrogel. Intracellular ice crystals were observed at 1.4 ms, with intracellular ice growth completing at 1.5 ms. Ultimately, at 22.5 ms, the hydrogel cracked due to thermal stress accumulated during the rapid cooling process. Although the hydrogel prepared using pure alginate reduced the size of the ice crystals, it was unable to prevent intracellular ice production in oocytes.

Intracellular ice crystals during high-speed quench cooling of alginate hydrogel-microencapsulated oocytes.
Figure 6b illustrates the vitrification process of antifreezing alginate hydrogel-encapsulated oocytes during quench cooling. At 3.7 ms, punctate ice crystals generated in the hydrogel and diffusively propagated, resembling the ice crystals in the VS2 group. By 4.4 ms, the ice crystals covered the entire field. Unfrozen solution in the interstices led to partial vitrification. At 9.3 ms, numerous black spots appeared in the hydrogel, possibly caused by solute precipitation, with no significant changes in the entire system.
Finally, cracks emerged in the hydrogel due to thermal stress. Importantly, no intracellular ice occurred throughout the process, demonstrating the successful vitrification of antifreezing alginate hydrogel-encapsulated oocytes using high-speed quench cooling.
Warming of antifreezing hydrogel-encapsulated oocytes
The antifreezing hydrogel-encapsulated oocytes underwent high-speed quench cooling and warming, and the images of oocytes at a warming rate of 150°C/min are presented in Figure 7. Upon reaching −52.1°C, nearly all the cracks in the visual field disappeared, accompanied by a brightening of the entire field due to the melting of ice crystals within the hydrogel. At −33.0°C, extracellular ice crystals recrystallized, resulting in a darkening of the entire field. The recrystallization of extracellular ice occurred almost simultaneously, while the oocytes remained unchanged. By −9.9°C, both the extracellular field and the oocyte darkened due to devitrification.

Intracellular and extracellular ice crystals of oocytes at a warming rate of 150°C/min. Scale bar: 50 μm.
Upon reaching −6.0°C, both extracellular and intracellular ice crystals melted, initiating a brightening of the visual field. With the temperature continuing to rise, the oocyte underwent a significant change in volume. Using ImageJ, the projected area of the oocyte was measured. The results revealed ∼2.85-fold increase in oocyte volume during the warming process from 10.5°C to 37°C, filling the entire zona pellucida space with cytoplasm. This dramatic change in oocyte volume indirectly suggests that IIF during warming disrupted the membrane structure of the oocyte, resulting in lethal damage.
Discussion
Cryomicroscopy stands out as the primary tool for studying ice crystals during the cryopreservation of cells.23,24 However, conventional video cameras on cryomicroscopes typically capture images at frame rates of 25 or 30 fps, limiting their ability to capture the intricate details of ice crystal growth in cells. Such cameras can only depict the blackening of cells as they freeze. The cooling rate of conventional cryomicroscopy ranges from 0.01°C/min to 100°C/min, generally suitable for conditions of slow freezing but inadequate for achieving the high cooling rates required for vitrification. In our study, we enhanced the cooling platform to achieve an average cooling rate of 2821.58°C/min.
For comparison, the conventional method of directly placing a 0.25 mL straw into liquid nitrogen yields a cooling rate of ∼2500°C/min. 25 Using the high-speed quench cooling device, we simulated the process of oocyte vitrification freezing and warming. During warming, we observed the growth of intracellular and extracellular ice crystals, revealing the impact of thermal stress on extracellular ice crystals.
As depicted in Figure 5, with increasing solution concentration, ice crystals nucleate at specific heterogeneous nucleation points. As the concentration of the remaining solution increases, the growth of crystal nuclei slows down. Both the crystal formation process and the concentration increase of the remaining unfrozen solution continue until the unfrozen solution becomes highly viscous and transforms into a glassy state. 26
In the VS1, VS2, VS3, and VS4 groups, the crystallization zone coexisted with the vitrification zone, but the final volume fraction occupied by ice crystals varied for different solution concentrations. Complete vitrification was achieved in the VS5 group (16.5% DMSO +16.5% EG), with no ice crystal growth observed in both intracellular and extracellular environments. Furthermore, a negative correlation was identified between the cooling rate and the critical CPA concentration. The high-speed quenching cooling rate in this experiment was 2821.58°C/min, and the corresponding critical CPA concentration was 16.5% DMSO +16.5% EG. If the cooling rate of the cryostage can be further increased, the critical CPA concentration is likely to decrease accordingly.
Recent studies have explored the use of hydrogels for cryopreservation.27–29 Water within hydrogels can generally be categorized into three states: free water, weakly bound water, and strongly bound water. 30 Free water and weakly bound water exhibit minimal interaction with the hydrogel network, while strongly bound water can maintain a liquid state even at −100°C. The content of free water is generally much higher than that of intermediate and strongly bound water.
Consequently, the hydrogel matrix alone provides limited inhibition of ice crystal formation. To enhance the antifreezing properties of the hydrogel, low-molecular-weight CPAs such as EG, DMSO, and alginate were physically mixed. EG and DMSO are well-known permeating cryoprotectants capable of entering cells, reducing intracellular water loss, preventing cell damage from osmotic pressure changes, lowering the freezing point of the solution, and delaying or inhibiting ice crystal formation both inside and outside cells.
In addition, they increase solution viscosity, promoting the transition to a glassy state. 31 7.5% EG +7.5% DMSO +0.5% alginate hydrogel produced a similar effect to the CPA consisting of 12% EG +12% DMSO +0.5 M trehalose. This effect arises from the strong hydrogen bonds formed between EG, DMSO, and water molecules in the hydrogel, disrupting the lattice structure between water molecules and reducing free water content, thus inhibiting ice crystal formation and propagation. 32
Considering the large size and high water content of oocytes, which make them prone to ice crystal formation during cooling, a predehydration step using a 1 M trehalose solution was employed before encapsulation. The vitrification of oocytes is attributed to two main factors. First, antifreezing hydrogels composed of EG and DMSO inhibit membrane surface nucleation of oocytes induced by extracellular ice crystals. Second, prehydration of oocytes in a 1 M trehalose solution for 1 minute reduces osmotically active water, protecting cells from osmotic shock and lowering the likelihood of IIF. 33
Traditional Cryotop vitrification methods for oocytes usually involve two steps: equilibration in ES at room temperature for 15 minutes, followed by transfer to VS for 1 minute before plunging into liquid nitrogen. 34 This loading process takes a total of 16 minutes, potentially causing osmotic and toxic damage to oocytes.
In contrast, the vitrification method presented in this article significantly mitigates these concerns. Initially, oocytes are predehydrated for 1 minute, cross-linked in the antifreezing hydrogel for 3 minutes, and finally encapsulated oocytes undergo “solvent replacement” for 2 minutes, significantly shorter than the conventional Cryotop method (6 min vs. 16 min). Experimental results indicate proper inhibition of extracellular ice crystal formation during the cooling process, and predehydrated oocytes do not trigger IIF. This allows for a 50% reduction in the concentration of osmotic CPA while still achieving oocyte vitrification.
Ice crystal recrystallization is observed between −33.0°C and −6.0°C, attributed to the limited maximum heating rate of the silver block controlled by electric heating, which is only 150°C/min. This slow heating rate is a primary contributor to recrystallization, a critical phenomenon during cryopreservation that can lead to fatal damage to cells. 35 According to the Kelvin effect, small ice crystals melt at lower temperatures, and the released liquid water collects in larger ice crystals, subsequently freezing again. 36
Studies have shown that the recrystallization rate of mouse oocytes in 1.5 to 2 M EG increases four to five times for every five-degree rise in temperature, corresponding to an activation energy of 26 kcal/mole, capable of causing lethal damage to cells. 37 Bischof et al 38 achieved the vitrification of human skin fibroblast microdroplets with a volume of 1 μL using 3D printing technology. During the rewarming process, gold nanorod-induced laser heating technology was employed, achieving a heating rate up to 106°C/min. This approach successfully avoided recrystallization, resulting in a final cell survival rate of 95%. The addition of a laser warming module to the visualization system may prevent recrystallization, allowing observation of intact oocytes after rewarming.
Experimental confirmation of critical CPA concentrations revealed that the combination of hydrogel and CPAs can effectively reduce the overall CPA concentration. Our evaluation criterion, based on microscopic images to assess oocyte integrity, serves as an initial step. However, further activation and fertilization experiments are necessary to validate whether oocytes retain fertilization and developmental competence after thawing.
Conclusion
In this study, we employed a high-speed quench cooling device to meticulously examine the swift cooling and warming dynamics of oocytes. Our investigation delved into the impact of hydrogel encapsulation on restraining both intracellular and extracellular ice crystals. Notably, the high-speed quench cooling, operating at an impressive rate of 2821.58°C/min, revealed an absence of ice crystals both inside and outside oocytes when treated with a CPA formulation of 16.5% DMSO +16.5% EG. The critical CPA concentration necessary for vitrification was determined through visualization techniques.
Intriguingly, when oocytes were encapsulated in an antifreezing hydrogel at a low osmotic CPA concentration (2.4 M), no ice crystals were observed during high-speed quench cooling. This suggests that antifreezing hydrogel encapsulation holds promise as a method for vitrifying oocytes. While mouse oocytes serve as a pivotal model in fundamental research, the direct translation of this approach to humans necessitates further validation using human oocytes. Nevertheless, our findings provide valuable insights into the cryopreservation of human oocytes.
Authors' Contribution
S.Y.Z. and X.L.Z. designed conceptualization and methodology; S.Y.Z, Y.Q.Z., and X.L. performed validation, formal analysis, investigation, and data curation; X.L.Z. provided resources, supervision, project administration, and funding acquisition; X.L. contributed to writing—original draft preparation; X.L. and X.L.Z. assisted with writing—review and editing. All authors have read and agreed to the published version of the article.
Footnotes
Author Disclosure Statement
The authors declare no competing interests.
Funding Information
This research was funded by the Clinical Research Plan of SHCD (SHDC2020CR3077B) and the National Natural Science Foundation of China (51376132).