
Abstract
Background:
Voltage-gated sodium (NaV) channels help regulate electrical activity of the plasma membrane. Mutations in associated subunits can result in pathological outcomes. Here we examined the interaction of NaV channels with cardiac arrhythmia-linked mutations in SCN2B and SCN4B, two genes that encode auxiliary β-subunits.
Materials and Methods:
To investigate changes in SCN2BR137H and SCN4BI80T function, we combined three-dimensional X-ray crystallography with electrophysiological measurements on NaV1.5, the dominant subtype in the heart.
Results:
SCN4BI80T alters channel activity, whereas SCN2BR137H does not have an apparent effect. Structurally, the SCN4BI80T perturbation alters hydrophobic packing of the subunit with major structural changes and causes a thermal destabilization of the folding. In contrast, SCN2BR137H leads to structural changes but overall protein stability is unaffected.
Conclusion:
SCN4BI80T data suggest a functionally important region in the interaction between NaV1.5 and β4 that, when disrupted, could lead to channel dysfunction. A lack of apparent functional effects of SCN2BR137H on NaV1.5 suggests an alternative working mechanism, possibly through other NaV channel subtypes present in heart tissue. Indeed, mapping the structural variations of SCN2BR137H onto neuronal NaV channel structures suggests altered interaction patterns.
Introduction
Voltage-gated sodium (NaV) channel mutations can cause dramatic alterations in cellular excitability leading to potentially life-threatening disorders. 1 In cardiac myocytes, NaV1.5 is the main NaV channel subtype responsible for heart muscle function.2,3 Consequently, mutations in this channel have indeed been associated with long QT syndrome (LQTS) 4 and Brugada syndrome (BrS). 5 However, it is now well established that NaV channels do not function in isolation but form membrane-embedded macromolecular complexes that include auxiliary proteins such as β-subunits,1,6–8 fibroblast growth factor proteins, calmodulin, and others.9–16 β-subunits are known to modify gating properties, regulate trafficking, and influence plasma membrane recruitment of NaV channels. 6 Four main β-subunit isoforms (β1–4) have been identified and despite a shared, single transmembrane-segment protein topology, each interacts distinctly with and exerts disparate effects on specific NaV channel subtypes (NaV1.1–1.9).6,17–19 All four β-subunits have been identified in atrial and ventricular cardiomyocytes, 2 and mutations have been implicated in cardiac disorders. 20
Potential disease-associated mutations have also been found in SCN4B (p.Ile80Thr) and SCN2B (p.Arg137His), encoding the NaV channel β4- and β2-subunit, respectively.1,21 These mutations were identified during genetic screening in two patients affected by familial atrial fibrillation and LQTS, respectively (www.ncbi.nlm.nih.gov/clinvar/). In this study, we investigated the functional and structural consequences of these mutations on NaV channel complex function, providing initial evidence of altered interactions.
Methods
Electrophysiological recordings in Xenopus laevis oocytes
DNA sequencing of human (h)NaV1.2, hNaV1.5 C373Y, hβ2, and hβ4 (Origene, USA) was confirmed by automated Sanger sequencing. 22 β-subunit mutants were generated using the Gibson assembly method. cRNA was synthesized using T7 polymerase (the mMessage mMachine Kit; Life Technologies, USA) after linearizing the DNA by restriction enzyme digest. hNaV channel constructs were expressed in Xenopus laevis oocytes (sourced from Xenopus one®) with or without a β-subunit (1:5 molar ratio) and electrophysiological recordings were taken 2–4 days post cRNA injection. Oocytes were maintained at 17°C in Barth's medium (96 mM NaCl, 2 mM KCl, 5 mM HEPES, 1 mM MgCl2, and 1.8 mM CaCl2, 50 μg/mL gentamicin, pH 7.6) and studied using the two-electrode voltage clamp recording technique (OC-725C; Warner Instruments, USA) with a 150 μL recording chamber. All data were filtered at 4 kHz and digitized at 20 kHz using pClamp 10 software (Molecular Devices, USA). The external recording solution used was ND100 (100 mM NaCl, 5 mM HEPES, 1 mM MgCl2, and 1.8 mM CaCl2, pH 7.6) and microelectrode resistances were 0.5–1.0 MΩ when filled with 3 M KCl. Recordings were performed at room temperature (∼22°C) and leak and background conductance, identified by blocking NaV1.2 and the tetrodotoxin (TTX)-sensitive NaV1.5 C373Y channel with TTX (Alomone Labs, Israel), was subtracted for all channel currents. Off-line data analysis was performed using Clampfit 10 (Molecular Devices), Excel (Microsoft, USA), and Prism 7 (GraphPad, USA).
Qualitative surface biochemistry
X. laevis oocytes expressing hNaV1.5, hβ4, hβ4-I80T, hβ2, hβ2-R137H were washed 2 × with ND100 and incubated with 0.5 mg/mL Sulfo-NHS-LC-biotin (Pierce, USA) for 1 h at 4°C. Oocytes were then washed 6 × in ND100 before lysis by sonication in 20 μL/oocyte Nonidet P-40 (NP-40, 1%) with protease inhibitor cocktail (Pierce). All subsequent steps were performed at 4°C. Lysates were gently shaken for 15 min and then centrifuged for 15 min at 14,000 rpm. The pellet was discarded and 40 μL of the supernatant was reserved as the total protein fraction. The surface fraction was generated by adding the remaining supernatant to 200 μL of MyOne™ Streptavidin C1 magnetic beads (Invitrogen, USA), raising the volume to 1 mL with NP-40, and then rotating overnight at 4°C. Beads were then washed 6 × with NP-40 and resuspended in 30 μL NP-40, after which the biotinylated protein was dissociated by addition of 1 × Bolt™ Sample Buffer plus Reducing Agent (Thermo Fisher Scientific, USA) and boiling at 95°C for 5 min. Protein concentrations were measured by bicinchoninic acid assay (Pierce) and samples were run on a Bolt 4–12% Bis-Tris Plus gel (Thermo Fisher Scientific), then subsequently analyzed by western immunoblotting. Membrane images were cropped to show relevant bands only.
Statistical analyses
Mean data points for fast inactivation time constants (τ), persistent current, and recovery from fast inactivation (RFI) were analyzed using a one-way analysis of variance. For data on normalized conductance-voltage and steady-state inactivation relationships presented in Table 1, unpaired Student's t-test against wild-type channel or wild-type β-subunit was used. Values in all cases reflect the mean and error bars reflect standard error of the mean. All analyses were carried out using Excel (Microsoft).
Gating Parameters of NaV Channels Without and in the Presence of β-Subunits and Mutants
N = number of experiments, d(x) = slope value.
p < 0.01 compared with WT channel by unpaired t-test.
p < 0.01 compared with WT β
SEM, standard error of the mean; WT, Wild-Type.
Recombinant protein production and crystallographic analysis
Human β4 C58A (residues 32–157) and β2 C55A (residues 30–153) were cloned into a modified pET28 vector.17,18 The β4-C58A/I80T and β2 C55A/R137H mutations were introduced using the QuikChange Kit from Agilent Technologies (USA). Both mutants were expressed at 18°C in Escherichia coli Rosetta (DE3) pLacI strains (Novagen, USA), induced at an OD600 of ∼0.6 with 0.4 mM IPTG, and grown overnight before harvesting. Cells were lysed through sonication in buffer A (250 mM KCl and 10 mM HEPES at pH 7.4), supplemented with 25 mg/mL DNaseI and 25 mg/mL lysozyme. After centrifugation, the supernatants were applied to a PorosMC column (Tosoh Biosep, USA), washed with buffer A plus 10 mM imidazole, and eluted with buffer B (250 mM KCl plus 500 mM imidazole pH 7.4).
The mutants were dialyzed overnight against buffer A and cleaved simultaneously with recombinant tobacco etch virus (TEV) protease. Next, the samples were run on another PorosMC column in buffer A, and the flowthrough was collected and dialyzed against buffer C (10 mM KCl plus 10 HEPES at pH 7.4), applied to a HiloadQ column (GE Healthcare, USA), and eluted with gradients from 0% to 30% buffer D (2 M KCl plus 10 mM HEPES at pH 7.4). Finally, samples were run on a Superdex75 (GE Healthcare) gel filtration column in buffer A. β4-C58A/I80T and β2-C55A/R137H were exchanged to 50 mM KCl plus 10 mM HEPES (pH 7.4), concentrated to 9.5 and 2.8 mg/mL, respectively using Amicon concentrator (3K MWCO; Millipore, USA), and stored at −80°C.
Crystallization, data collection, and structure solution
Crystals were grown using the hanging-drop method at 4°C. β4-C58A/I80T was crystallized in 0.1 M MES (pH 6.5), 15–17% (w/v) PEG 2000-MME and β2-C55A/R137H crystals were grown in 0.1 M HEPES (pH 7.0), 14% PEG 6000 using seeds from β2-C55A crystals at various dilutions. Crystals were flash frozen after transfer to the mother liquor supplemented with 30% glycerol. The datasets were collected at the beam line BL9-2 of the Stanford Synchrotron Radiation Lightsource and beam line 23-ID-D at the Advanced Photon Source (Chicago). Data were processed using XDS 23 and HKL3000. 24 Phases were obtained through molecular replacement through Phaser 25 using the available crystal structures of β4-C58A (4MZ2.pdb) and β2-C55A (5FEB.pdb). The structures were completed by manual model building in COOT 26 and refinement using Phenix. 27 Simulated annealing composite omit maps were calculated with CNS 28 software to verify the absence of residual model bias. All structure figures were prepared using PYMOL (DeLano Scientific, San Carlos, USA). Protein Data Bank (PDB) IDs for mutant β-subunits are 6VRR and 6VSV.
Circular dichroism spectroscopy
All proteins were diluted to 16 μM in 50 mM KCl, 10 mM K-phosphate, pH 7.6 supplemented with 5 mM β-mercaptoethanol. Spectra were measured from 200–260 nm in a Jacso J-810. The initial thermal melt of β4-C58A from 25°C to 95°C in a Peltier controlled sample chamber connected to a water bath showed highest secondary structural changes at 213 nm. For all subsequent melts, the signal at 213 nm was monitored as a function of temperature.
Results
We examined the effects of SCN4B, SCN4BI80T (β4I80T), SCN2B, and SCN2BR137H (β2R137H) on NaV channel gating. These mutations were chosen from the ClinVar database (www.ncbi.nlm.nih.gov/clinvar/), which contains many gene variants of unknown or unsubstantiated clinical significance. In this database, both variants are linked to LQTS 10, a cardiac electrophysiological disorder characterized by QT prolongation and T-wave abnormalities associated with tachyarrhythmias and syncope events that typically occur during exercise or emotional stress.29,30 Original data were submitted to ClinVar by Invitae, a genetic testing company based in California (USA). In this study, we exploit this information to investigate possible changes in SCN2B and SCN4B interaction with NaV channels, which may lay the foundation for future experiments in which a link with the clinical phenotype is investigated.
Electrophysiological analysis
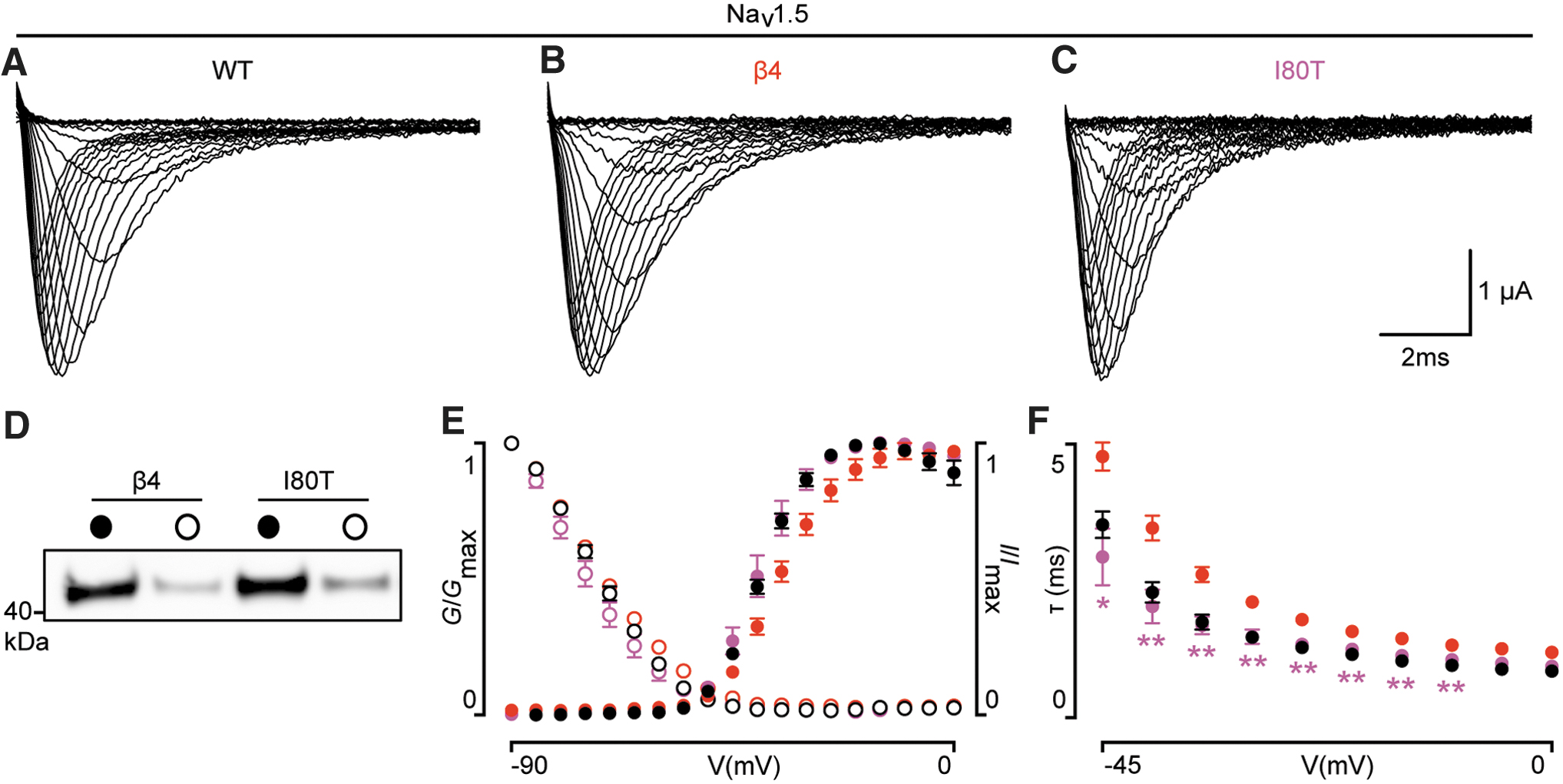
Since the clinical phenotype revolves around heart function, we used human (h)NaV1.5 as our model channel. 3 Similar to wild-type hβ4 and hβ2, both hβ4I80T and hβ2R137H express in Xenopus oocytes and translocate to the membrane, indicating that protein trafficking is not affected, at least in a heterologous Xenopus oocyte expression system (Figs. 1 and 2). To discern potential subtle effects of hβ4 and hβ2 on NaV channel function, we recorded and compared multiple gating parameters, such as the conductance-voltage (G-V) relationship, channel availability (I-V), RFI, and persistent current between hNaV1.5 alone or in the presence of hβ4 or hβ4I80T. Channel opening kinetics were not studied due to inherent technical limitations associated with electrophysiological recordings in Xenopus oocytes.

(
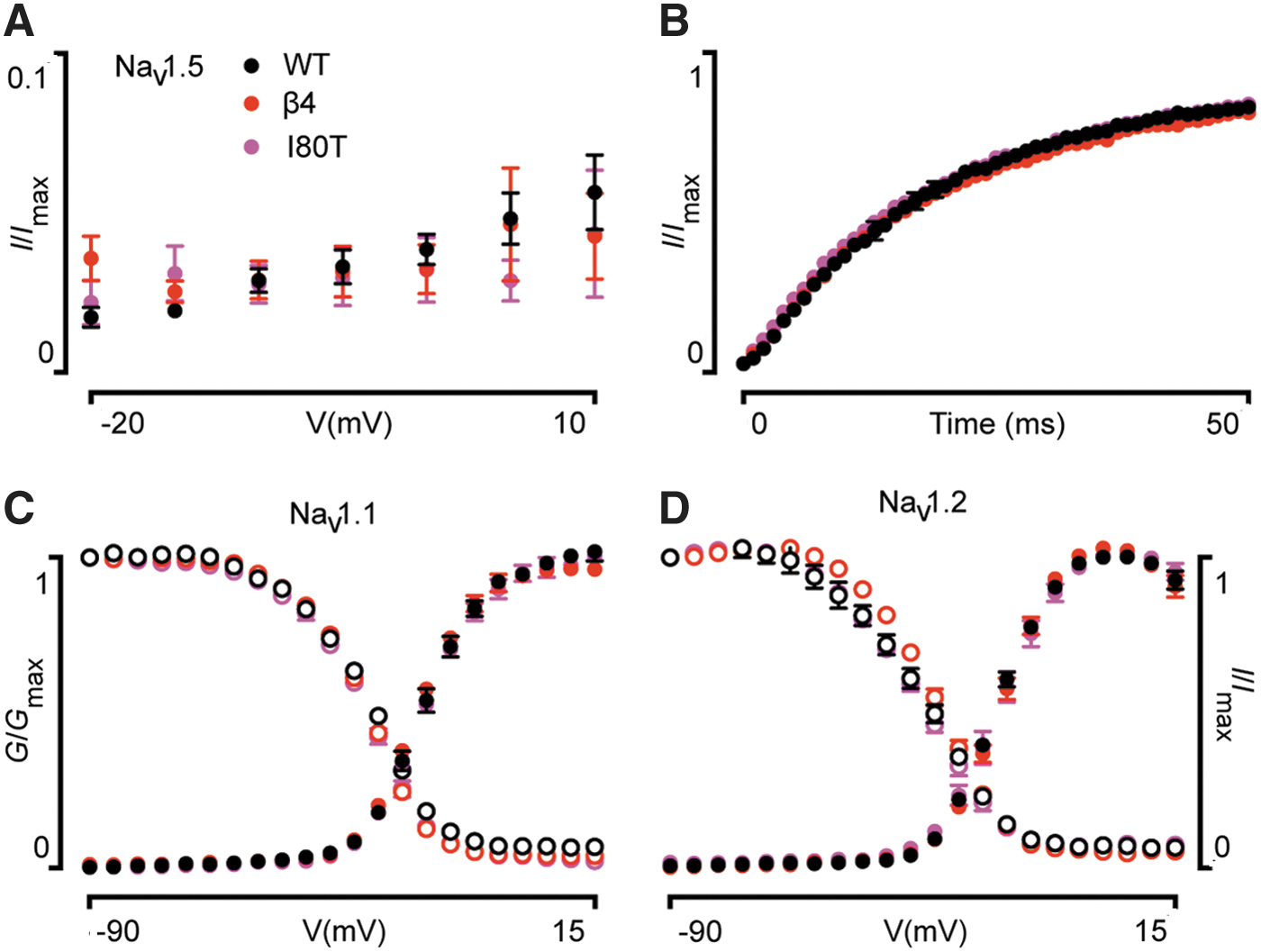
We found that the G-V relationship of hNaV1.5/hβ4I80T is shifted to more hyperpolarized potentials (±5 mV; p < 0.01) when compared with hNaV1.5/hβ4. A similar shift was seen in channel availability (±4 mV; p < 0.01). Another apparent effect of hβ4I80T emerged when fitting the current decay phase with a single-exponential fit. Over a 45-mV range, hβ4I80T caused NaV1.5 currents to inactivate significantly faster when compared with wild-type hβ4 (p < 0.05). Persistent current and RFI of hNaV1.5 were unaffected (Fig. 3). The G-V and I-V relationships of two neuronal NaV channel subtypes, hNaV1.1 and hNaV1.2, were not disrupted by the hβ4I80T mutation (Fig. 3). Markedly, hβ2R137H did not have any apparent effect on the tested hNaV1.5 gating parameters as compared with wild-type hβ2 (Fig. 2).
Crystallographic analysis
Since hβ2 and hβ4 can be crystallized,17,18 we prepared crystals for the hβ2R137H and hβ4I80T variants (PDB IDs are 6VRR and 6VSV). Both resulted in high-resolution structures of 1.45 and 1.65 Å, respectively (Table 2). Figure 4 shows a comparison of wild-type and hβ2R137H. The two structures can be superposed with an root-mean-square deviation (RMSD) of 0.79 Å for 90 Cα atoms (with Cα atoms of residues with double conformations not included). The R137H mutation results in multiple structural changes at distances >12 Å away from the mutation site. In wild-type hβ2, the Arg137 side chain is stacked against the side chain of Tyr128, making cation–π interactions. It also forms two hydrogen bonds with a water molecule involved in a hydrogen-bond network, including Ser64 and the Tyr128 main chain. In the hβ2R137H, these interactions are lost, resulting in a reorientation of the Ser64 side chain. Instead, His137 adopts a dual conformation and a 1.6 Å displacement of its main chain, which in turn propagates to several neighboring residues. This includes a near 10 Å shift in the side chain of Asn 28 , which makes a hydrogen bond with one of the His137 conformers, and large shifts of the Met130 side chain, His136, and Arg135. Overall, the R137H mutation results in significant changes in the local area around the mutation site. Although this seems at odds with the lack of effect of this mutation on NaV1.5 function (Fig. 2), it suggests that the structural changes do not disrupt the interface between hβ2 and NaV1.5. However, this mutation may lead to altered interactions with other NaV channel subtypes.
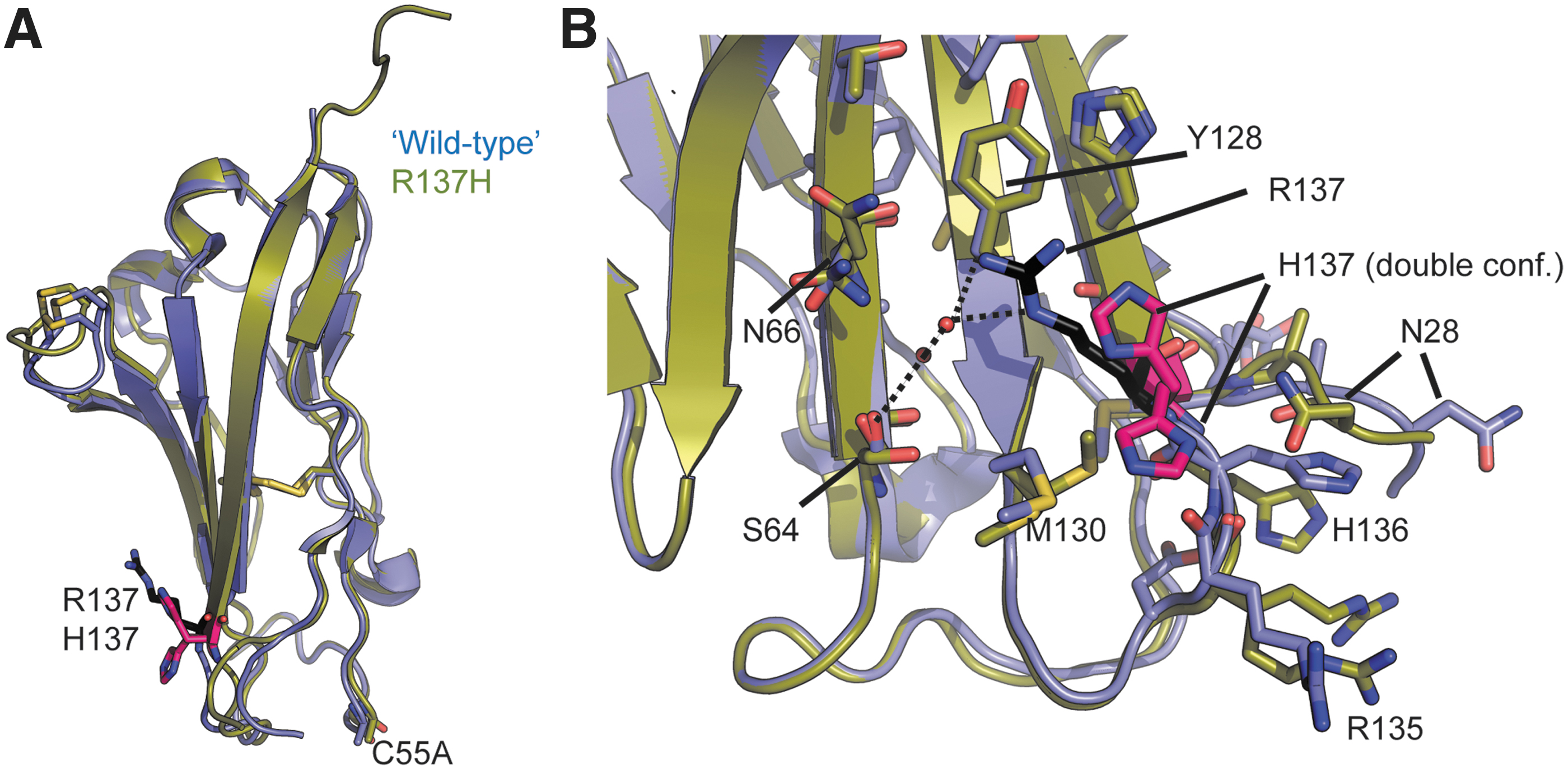
Crystallographic analysis of hβ2R137H. Wild-type hβ2 (containing the C55A mutation to facilitate crystallization
17
) is shown in blue, and the R137H mutant in green. R137 is shown in black sticks and H137 in pink.
Data Collection and Refinement Statistics
One crystal for each structure was used for data collection and structure determination.
Values in parentheses are for highest-resolution shell.
Value is for highest-resolution shell.
Using Procheck of CCP4 package.
The situation is very different for the hβ4I80T mutation. Wild-type and mutant subunit superpose with an RMSD of 0.48 Å for 105 Cα atoms (Fig. 5). In wild-type hβ4, Ile80 is buried within the hydrophobic core of the Ig-like domain, which consists of Trp68, Leu79, Val93, Leu103, and Ile101. Substitution by a hydrophilic residue would thus be predicted to be destabilizing. In addition, as Thr is smaller than Ile, the packing is less optimal; resulting in a change in the side chain of Leu103, which now adopts a dual side chain conformation. Importantly however, no structural changes are visible on the surface. As the surface of hβ4 would be interacting with NaV1.5, there is an apparent contradiction with the functional data, which clearly show an effect of the I80T mutation on the G-V relationship and steady-state inactivation (Fig. 1). However, as this protein crystallized at low temperature (4°C), and crystals were flash cooled with liquid N2, a possible temperature-dependent effect on the structure may not be observed. We thus postulate that the suboptimal packing in the hβ4 core can result in changes on the surface at elevated temperature, such as used in electrophysiological experiments or present in vivo.
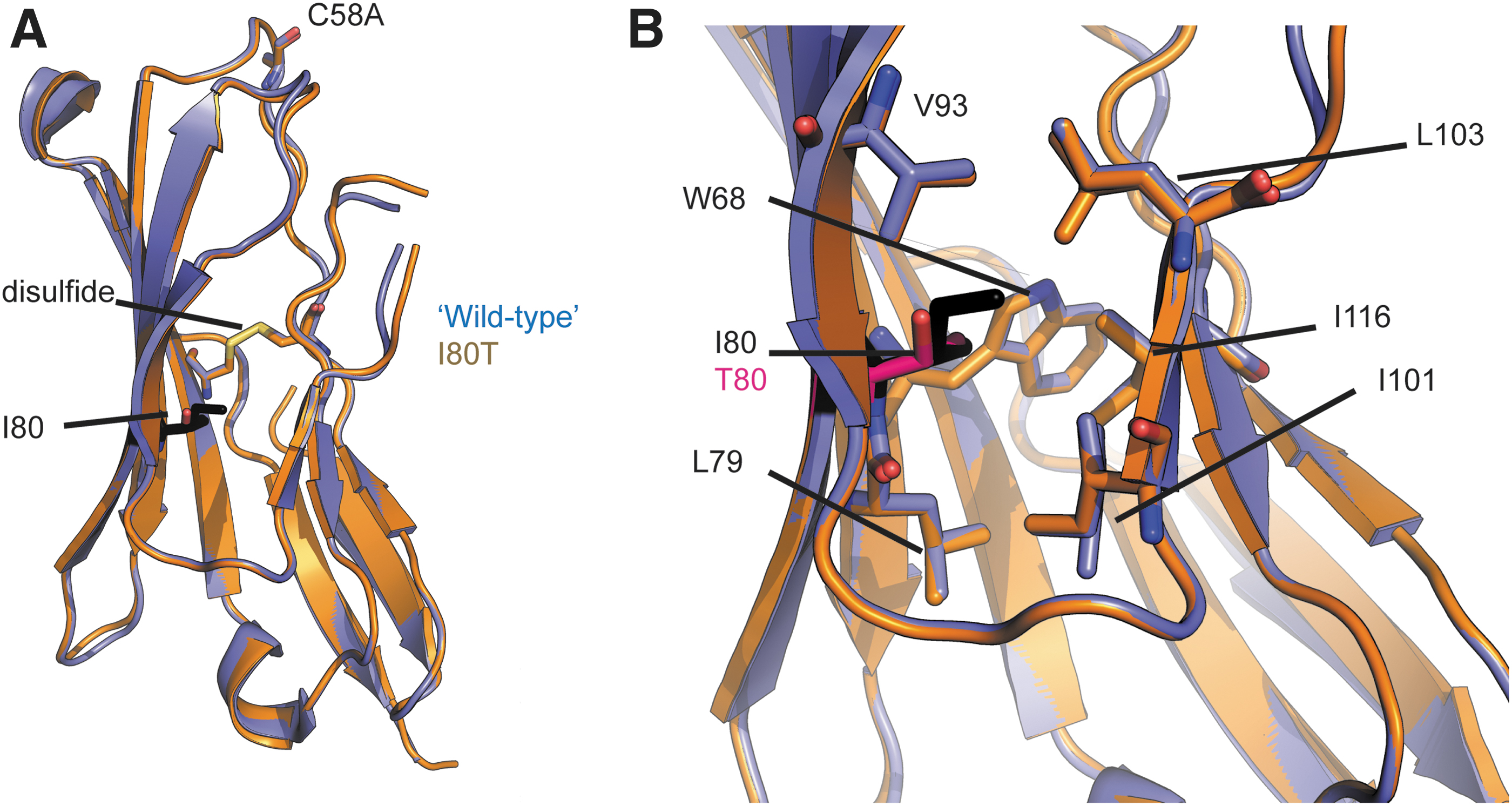
Crystallographic analysis of hβ4I80T. Wild-type hβ4 (containing the C58A mutation to facilitate crystallization) is shown in blue, and the I80T mutant in orange. I80 is shown in black sticks, and T80 in pink.
Protein stability
Amino acid substitutions may perturb protein folding. Although both β-subunit mutants still express to levels similar to wild type in Xenopus oocytes (Figs. 1 and 2), it is possible that these substitutions cause a thermal destabilization at higher temperatures. Such effect could explain how a mutant, which shows minimal structural changes in a crystallized form at low temperature, can still have a functional effect at physiologically relevant temperatures. We, therefore, prepared purified, recombinantly expressed extracellular subunits of hβ4, hβ2, and the two sequence variants, and utilized Circular Dichroism (CD) to measure thermal melting curves (Fig. 6). Monitoring the CD signal at 213 nm, we increased the temperature from 25°C to 95°C in increments of 0.5°C. Wild-type hβ4 displays a melting temperature (TM) of 61.1°C ± 0.3°C, whereas the I80T mutation results in a significant destabilization with a TM of 51.4°C ± 0.1°C (Fig. 6).
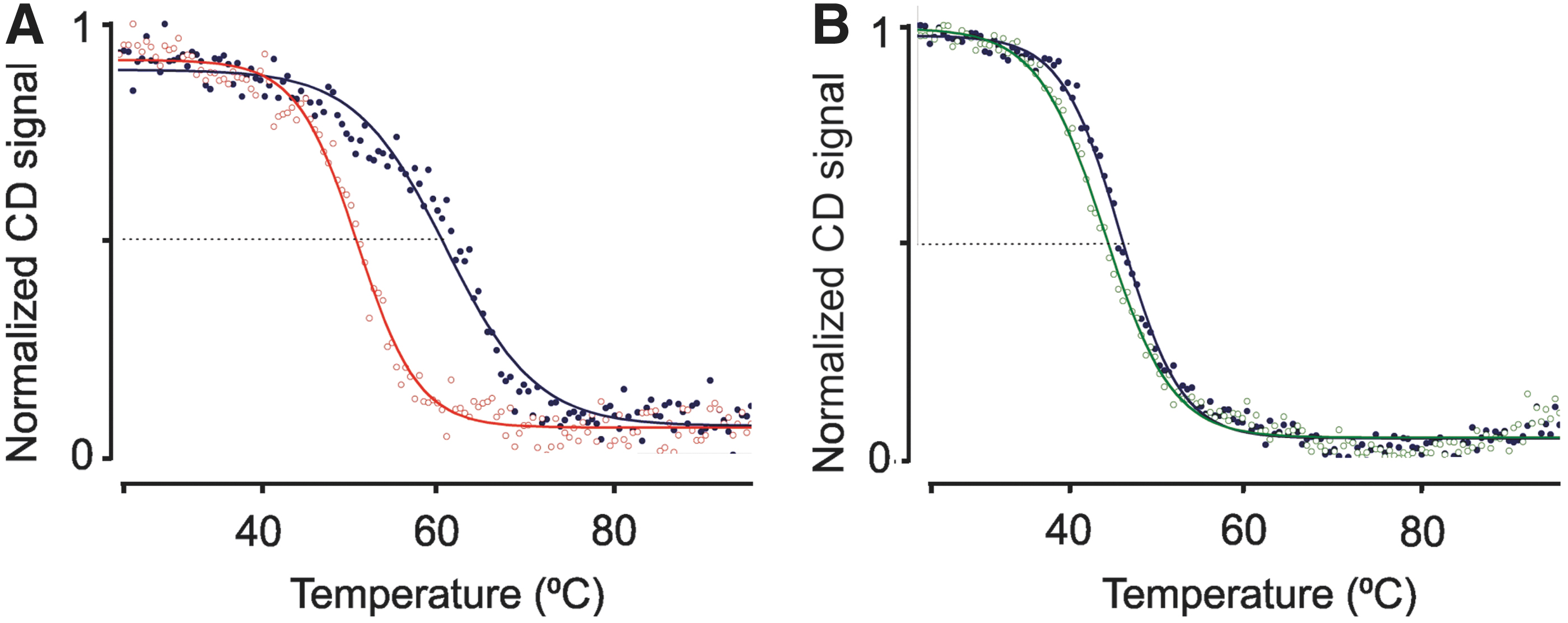
Thermal melting curves monitored through CD recorded at 213 nm.
Although the actual TM in the context of a native cell, under oxidizing conditions and in the presence of a NaV channel is unknown, our observation that the mutation has an effect on stability in our assay hints at possible structural changes at elevated temperatures. Although the TM is well above 37°C, a subtle degree of unfolding is already present at this temperature. Of note, the CD signal measures secondary structure, and it is thus conceivable that significant local structural perturbations, which do not affect secondary structure content, already occur at physiologically relevant temperatures, which would affect the interface between hβ4 and NaV1.5. Wild-type hβ2 is significantly less stable than hβ4, with a TM of 47.4°C ± 0.1°C. However, in this case, no significant impact on protein stability was observed for the R137H mutation (TM = 45.8°C ± 0.1°C).
Mapping β-subunit variants on cryoelectron microscopy structures
Next, we wondered whether the β-subunit mutations studied here are part of an interface with NaV1.5. A cryoelectron microscopy (EM) structure is available for NaV1.5 as well as crystal structures of cytosolic portions in complex with auxiliary proteins9,10,12–15,31 and also cryo-EM structures of the related NaV1.2, NaV1.4, and NaV1.7 channels, in complex with one or more auxiliary β-subunits.7,8,32 This includes structures of hNaV1.2 in complex with β2 (PDB 6J8E), hNaV1.4 in complex with β1 (PDB 6AGF), and hNaV1.7 in complex with β1 and β2 (PDB 6J8G). In each of the β2 complexes, a disulfide bond was observed with the pore-forming subunit. This corresponds to Cys910 in NaV1.2, a residue we predicted to form a disulfide bond with Cys55 in hβ2. 17 Although no cryo-EM structures have captured β4 in complex with a NaV channel, this subunit is also predicted to make a similar disulfide bond.17,18 However, as NaV1.5 does not have a Cys residue at the corresponding position, the interaction between β2/β4 with NaV1.5 cannot rely on this disulfide bridge.17,18,31
Figure 7 shows the structure of the NaV1.2-β2 complex (PDB 6J8E), illustrating that the interaction also involves several other residues that make extensive interactions with the Domain II pore-forming region of the channel. From these data, it is clear that the R137H variant is in a position to perturb this interaction. Although R137 is not directly located at the interface, the structural changes we observed for this mutation affect residues located at the β2-NaV1.2 interface, including Arg, 28 Arg135, and His136. Thus, the hβ2 R137H variant could perturb the interface with NaV1.2, and likely also closely related NaV channel subtypes expressed in the heart (i.e., NaV1.1–1.3). 33 Since hβ2R137H has no functional effect on NaV1.5 compared with hβ2, whereas hβ4I80T does, it seems unlikely that β2 and β4 interact with NaV1.5 similar to the NaV1.2-β2 complex. Indeed, NaV1.5 lacks the Cys equivalent to position 910 in NaV1.2, suggesting that the manner in which these two β-subunits interact with NaV1.5 is fundamentally different.17,31,34

Mapping the mutation sites on the NaV1.2-β2 cryo-EM structure.
NaV1.2 gating and β2R137H
Since neuronal NaV channel subtypes expressed in the heart 2 contain the Cys equivalent to position 910 in NaV1.2,7,17 the structural changes induced by the β2 R137H mutation may disrupt channel function. To test this hypothesis with links to available structural data, we used hNaV1.2 as our model channel, coexpressed with wild-type β2 or β2R137H. While we observe no changes in channel availability of hNaV1.2, the G-V relationship and fast inactivation in the presence of β2R137H differs substantially from that observed with wild-type β2 (p < 0.001) and is virtually identical to the condition when no subunit is present (Fig. 8).

Discussion
We investigated the functional and structural effects of two β-subunit variants, SCN4B (p.Ile80Thr) and SCN2B (p.Arg137His), found in patients with familial atrial fibrillation and LQTS 10. Our functional investigation showed a direct effect of the hβ4I80T, but not the hβ2R137H variant, on the function of hNaV1.5 (Figs. 1 and 2). However, structural investigation using X-ray crystallography revealed opposite results, with hβ2R137H causing significant structural changes and hβ4I80T resembling the wild-type protein under the crystallization conditions used (Figs. 4 and 5). A structural change does not necessarily have to cause a functional impact, especially if the perturbation is local and does not affect a protein–protein interface. The results presented here suggest that the region around Arg137 in hβ2 may not be involved in the interface with NaV1.5. However, the hβ4I80T results seem contradictory given that the absence of significant structural changes suggests that the interface with NaV1.5 should be unaffected.
To better understand this conundrum, it is important to consider a possible effect of temperature. Proteins will unfold, both locally and globally, with rising temperatures, and a mutation could affect this thermal stability.35,36 If any unfolding of the mutant occurs at physiological temperatures, and this affects a relevant interface, then the mutant is likely to have a functional effect. In this regard, the hβ4I80T caused a significant reduction in thermal stability (Fig. 6). This is not surprising, as our structural analysis showed that Ile80 is buried in a hydrophobic core, and substitution by a hydrophilic residue should be destabilizing. One could thus postulate that the mutation causes local changes in structure at or near the interface between hβ4 and NaV1.5 at physiologically relevant temperatures, leading to alterations in channel gating. Importantly, thermal melting curves obtained via CD only represent changes in secondary structure, so small structural perturbations may not be measured. Thus, although the melting temperature is above a physiologically relevant temperature, small local changes that do not affect secondary structure are still possible at physiological temperatures. In addition, the altered stability can affect the dynamics of hβ4, thus affecting its interactions with NaV1.5 indirectly.
Temperature-dependent effects have previously been reported as a result of mutations in NaV1.5. This includes amino acid substitutions in the EF-hand domain leading to thermal destabilization of this locus.37,38 Thermal destabilization induced by disease-causing mutations has also been described in other ion channel families.39,40 Since the hβ4I80T mutation was found in a LQTS patient, an increase in body temperature (e.g., fever or exercise) could elicit structural and functional changes in the cardiac NaV channel complex, thereby causing an increase in QT interval time and subsequently an increased risk to develop torsade de pointes and polymorphic ventricular tachycardia. Indeed, fever-related ventricular arrhythmia and sudden death have been associated extensively with BrS 41 ; however, few reports also describe fever-induced QT prolongation and ventricular arrhythmias in individuals with type 2 congenital LQTS. 42 As such, it is possible that the SCN4B p.Ile80Thr mutation could favor the appearance of an arrhythmic phenotype in concomitance with fever. We note that this remains a hypothesis, and it needs to be shown whether the mutation elicits small structural or dynamic changes at physiological temperatures.
Although the hβ2R137H variant did not appear to affect any of the electrophysiological properties of NaV1.5, mapping of the mutation on the NaV1.2-hβ2 cryo-EM structure showed that this residue can affect channel–subunit interactions. Although not directly involved in the interface, we found that the structural changes we observed affect residues directly participating in their interaction, including R135 and H136 (Fig. 7). Subsequently, hβ2R137H indeed restored hNaV1.2 function as if no hβ2-subunit was present when considering channel closure (i.e., the fast inactivation process) (Fig. 8). Since several neuronal NaV channel isoforms are expressed in the heart at functionally important locations,2,33 our result hints at a contribution of this hβ2 variant to arrhythmia,43,44 despite the absence of functional effects on the main cardiac isoform, NaV1.5. From a clinical perspective, hβ2R137H was found in a patient with familial atrial fibrillation and since intrinsic and extrinsic cardiac innervations are pivotal in determining physiological modulations in cardiomyocytes, 45 it may be feasible that this mutation can modify neural activity in the heart.
Finally, it is worth noting that the extracellular Ig domain of β-subunits can participate in homo- and heterophilic cell adhesion processes, which may have implications for cardiomyocyte function. 34 Combined with previous work,17,18,30,31,34,46,47 it is unlikely that the Ig domains of hβ2 and hβ4 interact with NaV1.5 in the same way that they do with other NaV channel subtypes, since NaV1.5 does not have a free Cys residue on the domain II extracellular loop.17,31 As such, the exact interface of hβ2 or hβ4 with NaV1.5 remains to be described, but likely does not involve hβ2 residue R137 or the neighboring residues whose positions are affected by the R137H mutation. Ig domains of hβ4 (and presumably hβ2) can form both cis and trans cell adhesion contacts and some of the same amino acids involved in binding to the NaV channel also contribute to these cis interactions. 34 Therefore, hβ2 and hβ4 Ig domains that associate with NaV1.5 may be involved in crosslinking of associated channels suggesting the involvement of higher-order assemblies in potentially causing cardiac defects and illustrating the complexities in linking genetic data to patient phenotype.
Footnotes
Author Contributions
F.B and F.V.P. conceived the experiments, analyzed data, and wrote the article. J.D.W., L.C., and A.S. helped write the article and interpret (clinical) data. S.D. performed all crystallographic and thermal melt experiments. J.P.L. performed all electrophysiology and biochemical experiments. All coauthors have reviewed and approved of the article before submission.
Acknowledgments
We acknowledge the staff at the Stanford Synchrotron Radiation Lightsource beamline BL9-2, and the Advanced Photon Source beamline 23-ID-D.
Disclaimer
The article has been submitted solely to this journal and is not published, in press, or submitted elsewhere.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
J.P.L. was supported by the James H. Gilliam Fellowships for Advanced Study through the Howard Hughes Medical Institute. F.B. received support from the National Institutes of Health (1R01NS091352). F.V.P. received support for this study by a grant from the Canadian Institute for Health Research (CIHR, PJT-148632). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the article.