
Abstract
The improvement of drug accessibility needs to be promoted by both drug innovation and drug imitation. Given the current situation in China where both the quantity of drug innovation and the quality of imitation need to be improved, China’s drug patent linkage system should primarily aim to promote competition in the drug market and take the elimination of potential competition-restricting effects as the basic orientation for improvement. NMPA and CNIPA can address the issue of “evergreen patents” by conducting substantive reviews of patent information and punishing deliberate improper registration of patent information. To prevent the abuse of the approval waiting period system, in substantive law, it is necessary to further clarify the constitutive elements of liability for damages caused by the abuse of the approval waiting period system; in procedural law, parties initiating litigation (or requesting an adjudication) may be required to provide guarantees. Regarding the issue of reverse payment settlement agreements, it is necessary to add provisions of the expiration of the exclusivity period for the first generic drug and the record review of reverse payment settlement agreements. For reverse payment settlement behaviors that harm social public interests, the people's procuratorate shall initiate civil public interest litigation.
I. INTRODUCTION
In July 2021, the National Medical Products Administration of China (hereafter referred as to the “NMPA”) and the China National Intellectual Property Administration (hereafter referred as to the “CNIPA”) jointly issued the “Implementation Measures for the Early Resolution Mechanism of Drug Patent Disputes (Trial)” [hereinafter referred to as the “Measures (Trial)”]. Additionally, the CNIPA released the “Administrative Adjudication Measures for the Early Resolution Mechanism of Drug Patent Disputes,” and the Supreme People’s Court promulgated the “Provisions on the Application of Law in the Trial of Civil Cases Concerning Patent Disputes Related to Drug Applications for Registration” (Judicial Interpretation [2021] No. 13) (hereinafter referred to as Judicial Interpretation [2021] No. 13). These three pivotal documents signify the initial establishment of drug patent linkage system in China. The drug patent linkage is an intricate institutional framework system, with different perspective and diverse focus, which can explain why the plethora of theoretical research contend different viewpoints on its optimization. Broadly speaking, academic perspectives on this topic in China can be categorized into three main streams: the first focuses on administrative and judicial efficiency, proposing improvements aimed at enhancing the efficiency of both administrative and judicial processes 1 ; the second emphasizes interest balancing, advocating that the optimization of the system should strike a balance between protecting drug patent rights, fostering the development of generic drugs, and enhancing drug accessibility 2 ; and the third takes a comparative law analysis approach, seeking to improve drug patent linkage system in China by drawing insights from comparable legal systems. 3 From a functionalist perspective, the formation and interpretation of legal norms transcend mere reliance on their initial content and logical deduction, instead necessitating attention to their societal implications. Within this analytical framework, the transplantation and enhancement of the drug patent linkage system must be steered by the fulfillment of its intended functionalities. Therefore, commencing from the fundamental essence of drug patent linkage and grounded in local realities, this article elucidates the functional orientation of the drug patent linkage system in China. It further delves into potential value deviations that may arise during its operation and ultimately offers pertinent recommendations for institutional refinement.
II. THE HISTORY AND STRUCTURE OF DRUG PATENT LINKAGE SYSTEM IN CHINA
A. The history of drug patent linkage system in China
Article 18 of the “Measures for the Administration of Drug Registration” (2007) is generally recognized as the “embryonic form” of China’s drug patent linkage system. However, this provision did not mention the relationship between patent disputes resolution and the approval of drug registration for market entry, leading to some wavering in the attitude of the former China Food and Drug Administration (hereafter referred as to the “CFDA”) toward this issue. In a document “Opinions on Intellectual Property Issues Related to Mannan Peptide,” CFDA stated that “the conditions for drug registration are safety, effectiveness, and controllable quality, and patent examination is not required for drugs.” However, in practice, when patent holders raised objections based on patent disputes, CFDA often adopted the approach of suspending the approval process. 4
In 2008, Article 69(5) of the Patent Law of the People’s Republic of China (hereinafter referred to as the “Patent Law”) introduced the “Bolar Exception” clause, explicitly outlining that the manufacturing, utilization, and importation of patented drugs or medical devices solely for the purpose of providing necessary information for administrative approval, as well as the exclusive manufacturing and importation of such patented products for these specific purposes, shall not constitute an infringement of patent rights.
In 2017, CFDA released the “Policies on Encouraging Innovation in Drugs and Medical Devices and Safeguarding the Rights and Interests of Innovators (Draft for Soliciting Opinions).” This document explicitly advocated the establishment of a drug patent linkage system and clarified that upon the patent holder initiating legal action in response to the certification of a generic drug manufacturer, CFDA shall establish a 24-month approval stay-period. This momentous step heralded the commencement of China’s journey toward establishing a comprehensive drug patent linkage system.
On January 15, 2020, China and the United States jointly inked the “Economic and Trade Agreement between the Government of the People’s Republic of China and the Government of the United States of America,” explicitly mandating the establishment of an “Effective Mechanism for Early Resolution of Patent Disputes” in China. In 2020, the “Measures for the Administration of Drug Registration” underwent a revision, resulting in the deletion of the original “Article 18.” Additionally, the same year witnessed the introduction of a “drug patent linkage” clause in Article 76 of the revised Patent Law. This clause outlined that during the process of drug listing review and approval, if a patent dispute ensues, the involved parties have the option to initiate legal action. Furthermore, NMPA is authorized to determine, based on the judicial outcome, whether to temporarily suspend the approval for market entry within a designated timeframe.
In July 2021, the sequential implementation of three pivotal documents mentioned above heralded the nascent establishment of a comprehensive drug patent linkage system in China. On January 18, 2023, the Supreme People’s Court delivered its definitive ruling in the landmark litigation case involving drug patent linkage, specifically resolving the dispute between Chugai Pharmaceutical Co., Ltd., and Wenzhou Haihe Pharmaceutical Co., Ltd., over whether a particular drug fell within the scope of patent protection. 5
B. The structure of drug patent linkage system in China
The drug patent linkage system in China comprises at least two integral aspects. First, it involves the functional interconnectedness between NMPA, CNIPA, and the court. Second, it encompasses the procedural linkage between the assessment and approval of generic drug registration and the settlement of patent disputes. By referencing Article 76 of the Patent Law and the stipulations outlined in the Measures (Trial), the drug patent linkage system in China can be dissected into three distinct components.
The first part is the fundamental institutional framework which provides opportunities for pioneer drug manufacturers to solve the dispute before the generic drug enters market and encompasses a disclosure system for drug patent information. Article 76(1) of the Patent Law explicitly stipulates that during the process of drug registration, parties involved may petition the courts to adjudicate whether the technical solutions pertaining to the drug under consideration fall within the scope of protection afforded by another party drug patent. Prior to the establishment of Article 76(1) of the Patent Law, patent disputes that emerged during the registration and approval process for drugs could solely be addressed through patent infringement lawsuits. Owing to the existence of the “Bolar Exception” clause, a unique provision within patent law, a situation may arise where, despite the technical solution of a generic drug falling within the scope of protection of pioneer drug patent, the court may judge that the generic drug manufacturer did not commit patent infringement. 6 It is noteworthy that merely determining whether a technical solution falls within the scope of protection of another’s drug patent does not automatically establish patent infringement liability. This indicates that China has not adopted the “artificial infringement” system practiced in the United States. Consequently, patent holders can only initiate confirmation actions, rather than seeking compensation for any infringement. Regarding the disclosure system for pharmaceutical patent information, Articles 2–4 of the Measures (Trial) mandate patent holders to register their pertinent drug patent details, on the China Listed Pharmaceutical Patent Information Registration Platform. The registration of pharmaceutical patent information by patent holders serves as the fundamental basis for safeguarding drug patents prior to generic drug entry. This registration ensures that relevant patent details are properly documented and considered. Contrary to existing patent information disclosure platforms, the pharmaceutical patent information registration platform adopts a drug-specific approach to disclosing pertinent patent details, thereby promoting a fair and transparent environment for the generic drug market.
The second part is the core procedural system that encompasses the certification of the generic drug, challenging listed patent, and approval waiting periods. According to Article 6 of the Measures (Trial), the certification of the generic drug system requires generic drug manufacturers to make certifications regarding each patent for the pioneer drug when submitting their registration applications for market approval. These certifications fall into four categories: (1) there is no relevant patent information; (2) the pioneer drug patent expired or was invalidated, or the generic drug manufacturer has obtained a license; (3) the generic drug manufacturer promises not to market the generic drug until the relevant patent of pioneer expires; and (4) the pioneer drug patent should be declared invalid or that the technical solution of the generic drug does not fall within the protection scope of the relevant patent. The patent challenging system is stipulated in Article 7 of the Measures (Trial): if patent holders or interested parties have objections to the fourth category of certification, they are required to file lawsuits with the courts or administrative adjudication with CNIPA within a specified period. Finally, Articles 8–9 of the Measures (Trial) establish the approval waiting period system. Upon receiving copies of case-filing notification from the court or the acceptance notice from CNIPA, NMPA temporarily suspends the approval process for generic drug market entry. NMPA will reinstate the approval process and render corresponding decisions in accordance with the judgment or adjudication received, with a maximum timeframe of 9 months. If the designated waiting period surpasses this 9-month limit, NMPA shall proceed to transfer the approval process to the next stage.
The third-party pertains to the supporting regulation, particularly the exclusivity period system granted to the first generic drug applicant. As stipulated in Article 11 of the Measures (Trial), in situations where the manufacturer of the first generic drug submits a certification asserting the invalidity of the pioneer drug patent, and subsequently, the patent is ultimately declared invalid, the NMPA shall impose a 12-month moratorium on the approval of marketing for any subsequent generic drug of the same variety, commencing from the date the first generic drug obtains marketing approval. The operational logic of the drug patent linkage system in China is illustrated in Figure 1.
III. THE FUNCTION ORIENTATION OF DRUG PATENT LINKAGE IN CHINA
The establishment of any system ultimately aims to address practical issues, thereby manifesting its value and guiding its improvement efforts. The design of China’s drug patent linkage system requires drawing on beneficial foreign experiences while standing on China’s national conditions, answering China’s questions, and summarizing China’s experiences. During the localization of the drug patent linkage system in China, concerns have been raised regarding its legitimacy from a theoretical perspective. Critics argue that this system could potentially hinder the registration of generic drugs because of patent-related dispute issues, favoring drug innovation but not necessarily leading to a reduction in drug prices. As a result, they contend that the system provides limited benefits in terms of protecting individuals’ lives and health rights. 7 Furthermore, some critics also argue that patent holders could exploit the drug patent linkage system to repeatedly delay the market entry of generic drugs, effectively extending their market monopoly and exclusive rights. 8 Supporters of the drug patent linkage system, however, contend that it effectively balances the interests of patent holders, generic drug manufacturers, and drug users, thereby adhering to the principles of fairness and justice. 9 The divergence in these positions actually reflects the value conflicts among drug patents, generic drug development, and the public’s health rights underlying the drug patent linkage system. Law is not only a system of norms but also a system of values. Therefore, it is necessary to delve deeper into the value functions upheld during the localization of drug patent linkage as a basic guide for system improvement. Considering the intricate relationship among drug access, drug R&D, and the current landscape of drug innovation in China, it is evident that both drug innovation and generic drug production hold significant importance. Promoting competition in the drug market is the functional orientation of drug patent linkage system in China.
A. The relationship between drug accessibility and drug R&D
The academic debate mentioned above has reached a basic consensus: the public’s access to drugs holds a priority position. In the long term, with the constant evolution of diseases that affect human life and health, the development of new drugs becomes indispensable in conquering these illnesses and safeguarding human health. From a utilitarian standpoint, enhancing intellectual property protection has the potential to stimulate technological innovation and optimize benefits for the greatest possible number of individuals. Therefore, the protection of drug patent rights, serving as an incentive mechanism for drug innovation, is inherently linked to the well-being and health of drug users. Nevertheless, the exorbitant prices of monopolistic drugs pose a substantial barrier to medication accessibility for patients. According to Locke’s theory of property rights, it is implied that when property rights hinder the survival of the poor, those rights ought to be relinquished. 10 Therefore, in situations where the patent rights held by patent holders clash with the life and health rights of drug users, and such conflict appears irreconcilable, drug patent rights typically need to yield in favor of protecting life and health rights in most cases. Generic drugs have become one of the most effective ways to lower drug prices.
However, the entry of generic drugs does not necessarily result in a substantial decrease in drug prices. According to some studies, as market concentration diminishes, the value of the substitution effect index for generic drugs also decreases. If the market concentration remains stable, the substitution effect index for generic drugs will also remain relatively unchanged. 11 Whether generic drugs can effectively lower drug prices depends, to a certain extent, on their quality. Without institutional incentives and safeguards, imitators tend to opt for lower-cost methods of drug replication, resulting in a redundant and low-quality supply of generic drugs that do not effectively contribute to lowering drug prices. It can be seen that despite their apparent opposition, drug accessibility, drug innovation, and drug imitation actually have a unified aspect: enhancing drug accessibility relies on increasing the number of drug innovations and improving the quality of drug imitation, and neither can be neglected.
B. The current status of drug R&D in China
The function of drug patent linkage ought to be aligned with the prevailing circumstances of domestic drug supply. In China, the drug supply side encounters a situation wherein both the quantity of drug innovation and the quality of drug imitation require enhancement. Based on the calculations conducted by the China National Pharmaceutical Industry Information Center, it was determined that in 2016, the generic drug market in China approximated 916.8 billion yuan. This significant figure represented approximately 60% of the overall drug market and a noteworthy 95% of the chemical formulation market, respectively. 12 From 2011 to 2016, the compound annual growth rate of the Chinese generic drug market was approximately 11.5%, but the growth rate has slowed in recent years. Although the development of domestic generic drug products in China has progressed swiftly, challenges of subpar quality and homogeneity persist. Research indicates that up until 2019, China had 2,603 domestic chemical generic drugs. However, the alarming statistic of 107,000 approval documents reveals that only 28% of these approvals correspond to generic drugs currently in production. Most products compete at a low-quality level. 13
The insufficiency of China’s drug innovation capabilities is evident in several ways. First, there exists a disparity between the level of R&D investment by domestic pharmaceutical companies and the global average. Although China’s drug R&D investment has consistently risen from 2014 to 2020, its proportion remains comparatively low in the global context of drug R&D funding (as illustrated in Fig. 2). 14 Second, the proportion of domestically developed innovative drugs is not significant. In 2023, among the new chemical drugs approved by NMPA, imported drugs comprised 69.2%. Among the 117 approved improved new drugs, 79 were imported, whereas only 38 were domestically produced. 15
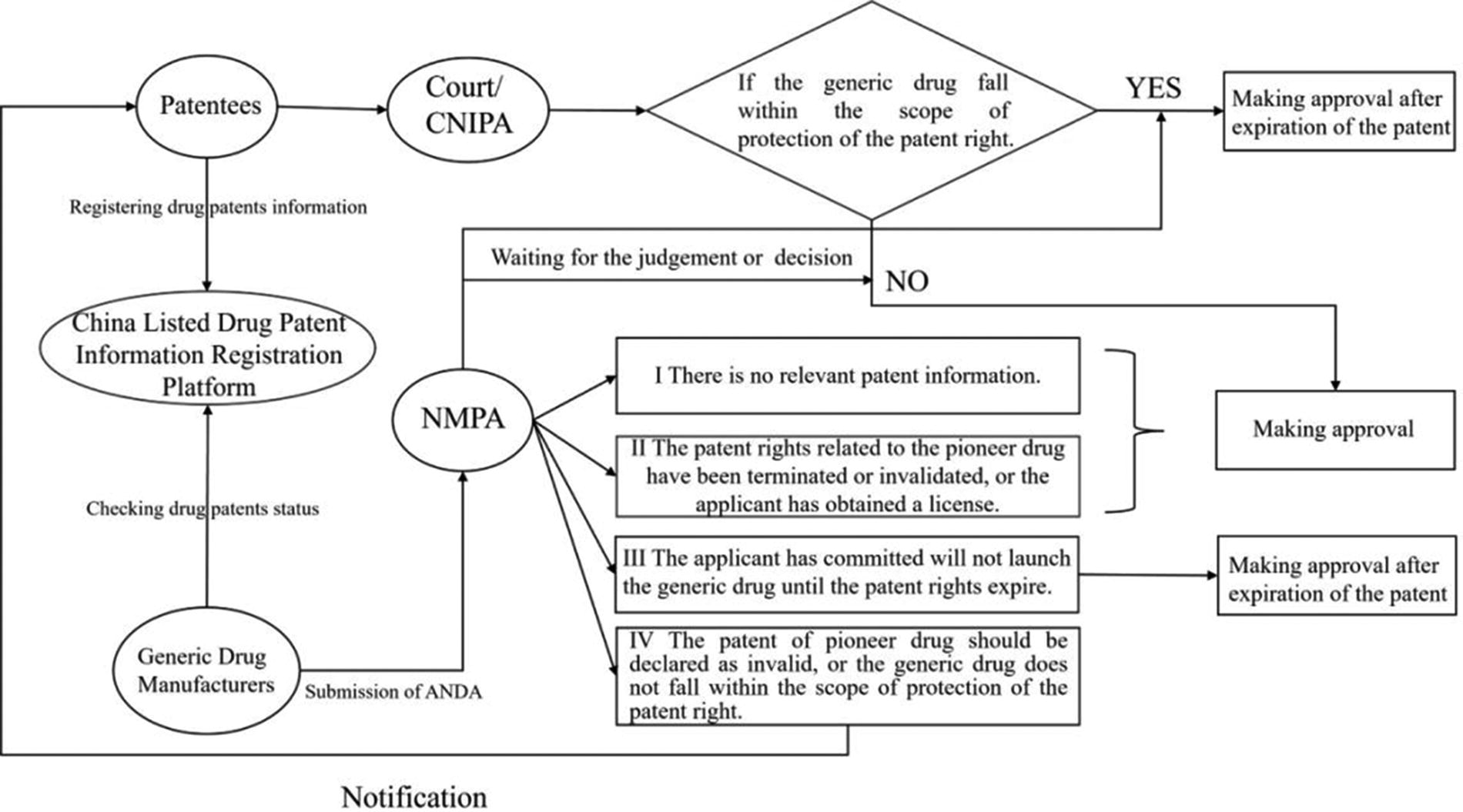
Schematic diagram of the operational logic of drug patent linkage system in China.

World Pharmaceutical R&D Investment vs. China Pharmaceutical R&D Investment (2014–2020).
Based on the reality of insufficient generic drug R&D capabilities and weak drug innovation capabilities, the drug patent linkage system in China should take into account both drug innovation and drug imitation, with the main function of promoting competition in the drug market.
C. The intrinsic mechanism of drug patent linkage in promoting competition
Before the implementation of the drug patent linkage system, patent disputes did not categorically prevent generic drugs from marketing. Nevertheless, this situation could often prove to be a double-edged sword. Despite successfully escaping from patent infringement issues during the registration process, generic drug manufacturers typically would face drawn-out patent infringement litigation and substantial compensation payouts once their products were released and sold.
The drug patent linkage system effectively tackles this problem by facilitating early resolution of patent disputes, thereby enhancing protection for patent holders. Additionally, it clarifies the status of rights and establishes a smoother and safer avenue for generic drug manufacturers to register their products, ultimately promoting a more equitable and efficient drug registration. 16 As stated by the United States Court of Appeals, District of Columbia Circuit in the case of Abbott Labs. v. Young: “Facing the classical question of the appropriate trade-off between greater incentives for the invention of new products and greater affordability of those products, Congress struck a balance between expediting generic drug applications and protecting the interests of the original drug manufacturers.” 17
In terms of its specific institutional components, the drug patent information disclosure system requires patent holders to register pertinent drug patent information. This, in turn, bolsters the transparency of drug patent information and diminishes the chances of patent infringement by generic drugs. The symbiotic relationship between these two systems effectively curtails the “transaction costs” associated with infringement liability. The patent challenge system enables patent holders to safeguard their legitimate rights and interests throughout the drug market entry approval process. Meanwhile, exclusivity period for the first generic drug system offers market exclusivity to the first successful challenger, thus motivating generic drug manufacturers to enhance their research and development capabilities and actively contest drug patents.
Indeed, following the establishment of the drug patent linkage system, the U.S. drug industry has witnessed a steady increase in investment toward drug R&D. A pivotal factor driving this trend is that the influx of generic drugs into the market, facilitated by the drug patent linkage system, has disrupted the monopoly profits enjoyed by drug patent holders. Consequently, to regain market dominance or monopolistic positions and secure profits, patent holders must ramp up their investments in the research and development of novel drugs. 18 This symbiotic approach ensures that both patent holders and generic drug manufacturers can prosper, fostering a virtuous cycle of innovation and competition in the pharmaceutical industry.
IV. THE POTENTIAL LIMITATION TO PHARMACEUTICAL COMPETITION IN DRUG PATENT LINKAGE
Guided by the functional orientation of fostering competition in the drug market and foreign legal experience, the implementation of the drug patent linkage system is not without challenges. Potential issues may arise and restrict market competition, including the utilization of “evergreen patents” to extend market exclusivity, the abuse of approval waiting periods to delay generic drug entry, and the employment of reverse payment settlement agreements to stifle competition.
A. Evergreen patents in the drug patent information disclosure system
“Evergreen patents” refer to new patent applications by drug patent holders that only involve marginal modifications to existing patents, without significant improvements in drug efficacy. 19 To prevent generic drugs from entering the market through the drug patent linkage system, patent holders continuously incorporate new patents, albeit unrelated to the core technology of the drug, into the drug patent information disclosure platform, effectively keeping the patents “evergreen.” For instance, when a generic drug manufacturer wishes to sell a generic version of a drug, it may challenge a specific patent (e.g., Patent No. 01). However, during the dispute resolution process, the patent holder may register a new patent (e.g., Patent No. 02) on the patent information disclosure platform. Even if the generic manufacturer successfully challenges Patent No. 01, it may still be unable to obtain marketing approval because of the existence of Patent No. 02. As a result, generic drugs face a perpetual “patent pool,” where patent holders can apply for multiple patents over several years, and each patent can independently block generic drugs from entering the market. 20 Canada, after adopting the drug patent linkage system, has been significantly troubled by this issue. Statistics show that in 2003, Health Canada only approved 16 new active substances, while patent holders added 103 patents to the Patent Register. 21
B. Abuse of the approval waiting period system
The abuse of the approval waiting period system occurs when patent holders, despite knowing that their patents have significant flaws or that the technical solution of generic drugs does not fall within the protection scope of their patent, still initiate litigation or request adjudication, maliciously triggering the approval waiting period. This can affect the registration process for generic drugs and delay their market entry. In general, suspending the registration process for generic drugs when patent disputes occur is a reasonable measure. Nevertheless, when this system is exploited by patent holders who deliberately initiate suits despite the high probability of their patents being invalidated, it transforms into a mechanism that unfairly prolongs the monopoly period enjoyed by the patent holders. For instance, in a series of patent disputes between Abbott Laboratories and Geneva Pharmaceuticals, Inc., the former filed five lawsuits in 1992, 1994, 1995, and 1996, spanning nearly 70 months, with only one successful outcome. 22 The Supreme Court of Canada, in the case of Apotex Inc. v. Merck Frosst Canada Inc. and Merck & Co. Inc., stated that “This prohibition takes effect automatically, without any consideration of the merits of the application; not even the ordinary requirements for an interlocutory injunction must be complied with”, “Under these conditions, and absent some prior indication to the contrary, I think it would be permissible for a generic producer to predict that either the patentee, the holder of a prior Notice of Compliance (NOC), or both, is likely to attempt to protect or prolong their as-yet exclusive rights for as long as possible by taking advantage of the procedure set out in the Regulations.” 23 Evidently, the abuse of the approval waiting period by patent holders unduly delays the market entry of generic drugs, imposing undue restrictions on drug competition.
C. Reverse payment settlement agreements
Reverse payment settlement agreements, commonly referred to as “pay-for-delay,” emerge as an inherent consequence of the drug patent linkage system. These agreements entail patent holders and generic manufacturers reaching a settlement wherein the generic manufacturer agrees to postpone market entry, thus delaying the commencement of generic drug sales and extending the duration of elevated drug prices. 24 What’s worse is that generic manufacturers may leverage the exclusivity period to negotiate settlements with patent holders, agreeing not to enter the market. During this exclusive time frame, the marketing of other generic drugs of the same type is prohibited, effectively extending the monopoly of the patent holder over the market. Data suggest that in the United States, reverse payment settlement agreements have led to an average delay of 17 months in the market entry of generic drugs, resulting in an estimated loss of $3.5 billion annually for consumers. 25 Reverse payment settlements provide generic manufacturers with certain guaranteed earnings, eliminating their incentive to compete in the market and disrupting drug competition, contrary to the legislative intent of the drug patent linkage system. The Supreme People's Court released ten typical anti-monopoly cases in 2022, among which the “Patent Infringement Dispute Case Involving the Reverse Payment Agreement for the Patent of Saxagliptin Tablets” is China's first “reverse payment settlement agreement case”, highlighting its actual existence within legal landscape in China.
V. INSTITUTIONAL IMPROVEMENT OF THE DRUG PATENT LINKAGE SYSTEM IN CHINA
A. Perfecting the disclosure system for drug patent information
A significant factor contributing to the rise of “evergreen patents” is the lack of a substantive review mechanism for drug patent information in the United States and Canada during that period. In a report, the Food and Drug Administration (hereafter referred as to the “FDA”) acknowledged its limitations in reviewing patent information because of a lack of expertise and emphasized that its resources should be primarily focused on assessing the drug application itself rather than patent assertions. 26 Consequently, FDA maintains a neutral stance on the patent information listed in the “Orange Book,” placing the onus of verifying patent information accuracy solely on the patent holders. In China, the Measures (Trial) do not explicitly outline whether NMPA should undertake a substantive review of drug patent information on the patent information disclosure platform. Article 4(2), merely stipulates that the responsibility for ensuring the authenticity, accuracy, and completeness of the registered information lies with the patent holders.
Given this, the improvement paths for the disclosure system for drug patent information are as follows: first, establish a substantive review mechanism for drug patent information. In addressing the issue of “evergreen patents,” South Korea made certain adjustments based on its national conditions when introducing the drug patent linkage system: patent information submitted by patent holders to the Ministry of Food and Drug Safety (hereafter referred as to the “MFDS”) must be directly related to the drug before it can be listed. 27 After receiving an application for the listing of an original drug, MFDS reviews whether the relevant patents meet the requirements for inclusion in the drug patent registration platform, particularly the relevance of the patents to the drug. The patent information that is not directly related to the listed drug is to be excluded to expedite the registration of generic drugs. 28 There is controversy over whether China should establish similar regulations. Supporters argue that substantive review helps screen out flawed patents and derivative patents, enabling innovative and practical patents to be granted. 29 Opponents believe that NMPA only has the authority to review the safety, effectiveness, and quality controllability of drugs and lacks the professional expertise and administrative resources to conduct a substantive review of patent information. 30 The obstacles mentioned by opponents are not unsurmountable. Even if NMPA finds it difficult to independently complete the review of patent information in the short term, it can still achieve this by establishing a collaborative liaison mechanism with CNIPA. Moreover, since both NMPA and CNIPA belong to the State Administration for Market Regulation, there are fewer obstacles to cooperation. Therefore, an option is to jointly establish a substantive review mechanism for drug patent information between NMPA and CNIPA.
Second, establish a supervision mechanism for drug patent information. In comparative law, the drug patent linkage system in South Korea has established an expedited procedure for deleting flawed patents from the drug patent registration platform: MFDS has the authority to delete or modify patents in the following circumstances: (1) where the patent holder withdraws the application; (2) where the relevant patent no longer meets registration requirements; (3) where a drug patent is registered by fraud or other improper means. This approach can be used for reference. 31 After the disclosure of drug patent information, NMPA and CNIPA should regularly conduct dynamic tracking of patent information and eliminate patents unrelated to the core technology of the drug. When feasible, the two departments can consider to collaborate by selecting qualified personnel to establish jointly establish a temporary internal agency specifically responsible for the review, registration, disclosure, and other matters related to drug patent information. For any relevant objections received pertaining to drug patent information, NMPA should notify the patent holders to verify and handle them in a timely manner. If the latter refuses to or fails to handle within the specified time limit, NMPA and CNIPA shall handle them according to their authority.
Third, in dealing with the intentional improper registration of patent information, it is necessary to clarify the legal responsibilities of such behaviors. Article 15 of the Measures (Trial) stipulates the adverse consequences that patent holders who commit improper registration should bear, but there are different theoretical opinions on the nature of the legal responsibilities borne by registrants. Some argue that administrative departments should solely impose administrative penalties to deter such behavior. 32 Others contend that, in addition to administrative penalties, those who improperly register should also bear civil responsibilities, including compensation for any potential damages caused by their actions. 33 In terms of constituent elements, the improper registration can only lead to losses for imitators when combined with the triggering of the “approval waiting period.” Therefore, there may be significant practical obstacles in establishing a causal relationship between improper registration alone and the losses suffered by imitators. Moreover, as previously discussed, the disclosure of drug patent information is closely related to drug competition and social welfare. Therefore, it is more consistent with the main function of the drug patent linkage to clarify that patent holders who intentionally and improperly register drug patent information should bear administrative responsibilities. In terms of the content of the responsibility, improper registrants may be subject to joint disciplinary action and they may not apply for registration of the same drug within 1 year.
B. Regulating the abuse of the approval waiting period system
Regarding the legal regulation of the abuse of the approval waiting period system, there are two predominant theoretical frameworks: substantive regulation and procedural regulation. First, patent holders who abuse approval waiting period system are liable for any potential harm inflicted on generic drug manufacturers. According to Judicial Interpretation [2021] No. 13, Article 12 stipulates that if a patent holder or an interested party knows or should know that the patent should be invalidated or that the technical solution of the generic drug does not fall within the protection scope of patent, but still initiates a lawsuit or requests administrative adjudication, the generic drug applicant may file a lawsuit for damages. Second, procedural regulation emphasizes that the initiation should be preceded by an application submitted by the patent holder, and NMPA will decide whether to suspend the approval process after conducting a substantive review of the application. Comprehensively speaking, a combined approach of substantive and procedural regulation is more appropriate, but there is still room for further discussion and optimization.
In terms of substantive regulation, further clarification is warranted regarding the constituent elements of liability for damages stemming from the abuse of the approval waiting period system. First, the subjective fault of the patent holder can be dissected into two distinct aspects. One aspect pertains to the knowledge of significant flaws in the drug patent or the realization that the technical solution of the generic drug does not fall within the protection scope of patent. The other aspect concerns the deliberate act of initiating litigation (or requesting adjudication) with the ulterior motive of impeding the approval process for generic drug entry approval. These two aspects, taken together, constitute the subjective fault of the patent holder in the context of abusing the approval waiting period system. Both are indispensable. Second, the amount of compensation for damages should be determined based on the actual losses of the generic drug manufacturer. According to the principle of compensation in civil law, the actual losses of the generic drug manufacturer are calculated based on the formula of “(the date when NMPA actually approved the generic drug entry − the date when NMPA should have approved the generic drug) × the average daily sales of similar drugs.” Some theorists argue that the amount of compensation for damages should include the profits obtained by the patent holder as a result of the abuse. It should be noted that the abuse of the “approval waiting period” by the patent holder does not fall within the scope of situations where the infringement proceeds should be returned as stipulated in Article 1182 of the Civil Code of People’s Republic of China and Article 71(1) of the Patent Law. Finally, the foreseeability rule can be used to determine the causality between the behavior of the patent holder and the losses of the generic drug manufacturer. The harm inflicted on generic drug manufacturers by abuse of the approval waiting period system is characterized as “pure economic loss.” This loss is marked by a significant degree of contingency and uncertainty, necessitating the utilization of specific instruments to establish necessity amidst this contingency. The suggestion by some scholars to borrow from legal experiences of Common Law System in determining pure economic losses is an intriguing one. The introduction of the “foreseeability rule” from contract law, in particular, can provide a valuable framework for assessing and managing such losses. According to the general social experience and wisdom of a rational person, if the corresponding damage is a usual consequence of the behavior, then the damage is foreseeable to the actor, and there is a causal relationship between the behavior and the damage. Based on the general principle of the foreseeability rule, it is foreseeable to a rational person in society that the abuse of the approval waiting period system by the patent holder will cause damage to the generic drug manufacturer. Therefore, there is considerable room for the application of the foreseeability rule in the determination of liability for damages arising from the abuse of the approval waiting period system.
Regarding procedural regulations, apart from NMPA conducting a substantive review of the application patent holder before deciding whether to suspend the approval process for generic drugs, it is also necessary to stipulate that the court (or CNIPA) may require the patent holder initiating a lawsuit (or requesting an adjudication) to provide guarantee for the following reasons. First, requiring patent holders to provide a guarantee is an indispensable measure for striking a balance between competing interests. Initiating a lawsuit (or requesting an adjudication) will postpone the time for generic drugs to enter the market, which may restrict the interests of generic drug manufacturers and social welfare. The existing substantive regulation as mentioned above cannot satisfy the protection of social welfare. Therefore, requiring patent holders to provide a guarantee can serve as a means of balancing interests. Second, requiring a guarantee serves as a warning to patent holders to exercise caution when initiating a lawsuit (or requesting an adjudication). By requiring patent holders to provide a guarantee, the court (or CNIPA) effectively gives patent holders an incentive to reassess the necessity of initiating a lawsuit (or requesting an adjudication). Compared with potential infringement suits that may arise afterward, the effect of providing a guarantee is more realistic and intuitive, which is more conducive to preventing patent holders from abusing procedural rights. Last, providing a guarantee will not impose an undue burden on patent holders. In reality, patent holders are typically well-established pharmaceutical companies with substantial financial resources, and thus, furnishing a guarantee will not pose a significant financial strain on them. In terms of specific rule-setting, the court (or CNIPA) can determine whether a patent holder should provide a guarantee based on the basic circumstances of the case, such as the number of times the patent holder has initiated a lawsuit (or requested an adjudication) against a particular generic drug, the preliminary evidence provided by the patent holder, and the impact on social welfare. The court (or CNIPA) can determine the amount of the guarantee by considering factors such as the potential financial losses that the lawsuit (or request an adjudication) may cause to generic drug manufacturers, as well as the potential increase in medication costs that could be passed on to the public.
C. Regulating reverse payment settlement agreements
First, in terms of establishing the invalidation regulations for the exclusivity period of the first generic drug, considering that the exclusivity period can induce parties to reach reverse payment settlement agreements, some scholars argue that the exclusivity period for the first generic drug has an insignificant effect on reducing drug prices and may even have a reverse incentive effect. Thus, it should be abandoned. 34 The other scholars believe that the invalidation causes for the exclusivity period of the first generic drug can be set to address the emergence of reverse payment settlement agreements. 35 Overall, the exclusivity period system aligns with the status of local drug R&D in China, aiming to incentivize generic drug development. Furthermore, considering that Article 11 of the Measures (Trial) already establishes an exclusivity period system, maintaining this system aligns more closely with the stability requirement of law, as outlined by the “Radbruch Formula”.
In U.S. law, the exclusivity period becomes invalid under the following circumstances: (1) where the first generic drug fails to enter within 75 days after obtaining marketing approval or within 30 months after submitting the marketing registration application; (2) where the first applicant withdraws the marketing registration application; (3) the first applicant amends or withdraws the certification for all of the patents with respect to which that applicant submitted a certification qualifying the applicant for the 180-day exclusivity period; (4) the first applicant fails to obtain tentative approval within 30 months; (5) the first applicant reaches an agreement with other applicants, patent license holders, or patent owners that violates antitrust laws; (6) all of the patents as to which the applicant submitted a certification qualifying it for the 180-day exclusivity period have expired. 36
In contrast, South Korean law specifies four causes for the invalidation of the exclusivity period for the first generic drug: (1) where the ruling to declare the patent invalid or the generic drug not falling within the protection scope of the patent obtained by is revoked or overturned; (2) where the first applicant fails to market the generic drug within 2 months of being eligible for marketing without good cause; (3) where the first applicant abuses market dominance, reaches monopolistic agreements, or carries out unfair trading practices; (4) where the applicant has obtained market exclusivity through fraud or other improper means. 37
China can draw upon the experiences of both the U.S. and South Korean laws to establish invalidation situation for the exclusivity period of the first generic drug: (1) where the first applicant fails to market the generic drug within 2 months of its marketability without justification; (2) where marketing approval for the first generic drug is revoked; (3) where the applicant has anticompetitive behavior by means of the exclusivity period system; (4) where the first applicant obtained market exclusivity through improper means.
Second, it is necessary to establish a submission and testing mechanism for reverse payment settlement agreements. Reverse payment settlement agreements effectively prolong the monopoly status of drug patents, weakening competition and undermining the protection of social welfare. Consequently, it is imperative to regulate such agreements procedurally. From a comparative perspective, the U.S. law requires that when a reverse payment settlement agreement is reached between a generic drug applicant and a patent holder during the drug marketing registration approval process, a copy of the agreement must be submitted to the Assistant Attorney General of the Antitrust Division of the Department of Justice and the Federal Trade Commission for registration within 10 days of its conclusion. 38 South Korean law dictates that the parties of the agreement must report the parties involved, content, and timing of the reverse payment settlement agreement to the MFDS and the Fair Trade Commission within 15 days of its conclusion. 39 Therefore, China can draw upon these foreign legal experiences and require patent holders and generic drug applicants to submit a copy of the reverse payment settlement agreement to NMPA and the National Antitrust Committee for registration within 10 days of its conclusion.
Third, it should be clearly stipulated that the procuratorates are empowered to initiate public interest litigation against reverse payment settlement behaviors. Article 60 of the Anti-Monopoly Law of the People’s Republic of China dictates that if an operator engages monopoly practices that harm public interests, the procuratorates may initiate civil public interest litigation. There have been controversies in the U.S. judicial practice regarding whether reverse payment settlement agreements violate anti-monopoly law. In the case of Louisiana Wholesale Drug Co., et al. v. Hoechst Marion Roussel, Inc., and Andrx Pharmaceuticals, Inc., the court held that the agreement between Hoechst Marion Roussel and Andrx Pharmaceuticals, where the former paid the latter US$40 million annually and the latter refrained from selling Cardizem CD and its generics in return, despite obtaining a marketing authorization, aimed to eliminate competition in the Cardizem CD drug market and constituted a horizontal monopoly agreement. 40 However, in Valley Drug Co. v. Geneva Pharms., the court suggested that patent monopoly is an exception to anti-monopoly law, and if the effect of a reverse payment settlement agreement falls within the scope of patent monopoly exemption, such agreement is not subject to anti-monopoly regulations. 41 In 2013, the U.S. Supreme Court, in order to unify judicial practice, clarified in FTC v. Actavis, Inc. that reverse payment settlement agreements should be subject to anti-monopoly regulations. 42 To enhance the safeguarding of public interests and establish a comprehensive regulatory framework alongside anti-monopoly administrative enforcement, it is imperative to oversee reverse payment settlement agreements via public interest litigation. When reverse payment settlement agreements hinder the public access to drugs and cause harm to public interests, the procuratorates can initiate civil public interest litigation in accordance with their authority.
VI. CONCLUSION
Throughout history and across cultures, the field of medicine has always been deeply rooted in ethics and morality, with drugs occupying a pivotal position. Patent protection serves as a powerful incentive for drug innovation, yet it also indirectly contributes to the high cost of medicines. The ethical and moral dilemmas arising from the implementation of the drug patent system are not only a challenge to the practical rationality of the law but also a questioning of its ideal rationality. Resolving the human rights dilemma in the drug patent system hinges on both the allocation of drug resources and the overall supply of drugs. In a country like China, where the level of drug imitation and innovation needs to be improved, there exists a substantial intersection between safeguarding drug innovation, advancing the research and development of generic drugs, and guaranteeing drug accessibility. Consequently, the objective of the drug patent linkage system in China ought to be the facilitation of competition within the pharmaceutical market. Potential issues that restrict competition, including evergreen patents in the drug patent information disclosure system, abuse of approval waiting periods, and reverse payment settlement agreements, should be the focus of attention in the development of the drug patent linkage system. First, NMPA and CNIPA can collaborate to review and supervise drug patent information to be listed on the platform, penalizing those who intentionally register improper drug patent information. Second, patent holders who abuse the approval waiting period for the purpose of delaying market entry of generic drugs should be held liable for compensation. The court (or CNIPA) may, as the case may be, require the patent holder who initiates a lawsuit (or requests an adjudication) against the patent challenging to provide a guarantee. Third, to tackle the problems of reverse payment settlement agreements, China can leverage insights from comparative law and implement a system that revokes the exclusivity period. Additionally, it should be stipulated that parties involved in such agreements shall submit the copies for review purposes. The procuratorates should have the authority to initiate civil public interest litigation against those agreements which undermine the public interest.