
Abstract
Background:
Movement disorders encompass various conditions affecting the nervous system. The pathological processes underlying movement disorders lead to aberrant synaptic plastic changes, which in turn alter the functioning of large-scale brain networks. Therefore, clinical phenomenology does not only entail motor symptoms but also cognitive and motivational disturbances. The result is the disruption of motor learning and motor behavior. Due to this complexity, the responsiveness to standard therapies could be disappointing. Specific forms of rehabilitation entailing goal-based practice, aerobic training, and the use of noninvasive brain stimulation techniques could “restore” neuroplasticity at motor–cognitive circuitries, leading to clinical gains. This is probably associated with modulations occurring at both molecular (synaptic) and circuitry levels (networks). Several gaps remain in our understanding of the relationships among plasticity and neural networks and how neurorehabilitation could promote clinical gains is still unclear.
Purposes:
In this review, we outline first the networks involved in motor learning and behavior and analyze which mechanisms link the pathological synaptic plastic changes with these networks' disruption in movement disorders. Therefore, we provide theoretical and practical bases to be applied for treatment in rehabilitation.
Impact statement
The pathological processes underlying movement disorders lead to aberrant synaptic plastic changes, which in turn alter the functioning of large-scale brain networks. This review article arises from the analysis of these topics, which cover a wide translational range of knowledges. The need to better understand the complexity subtending the expression of movement disorders in terms of plasticity changes and network de-arrangements is extremely actual. In neurorehabilitation, these theoretical and practical notions should not be ignored. In this concern, this article provides relevant information for both neuroscientists and clinicians to be applied for the study and the management of movement disorders.
Introduction
Motor behavior results from complex neural interactions, which are essential to decode environmental and motivational inputs for planning action schemata to be sent to the motor system (Goetz and Shackelford, 2006). Motor behavior continuously adapts itself to environmental, ever-changing internal and external conditions (Walker, 1999). This is possible because neurons are organized into microscale networks and large-scale networks (Bressler and Menon, 2010; Edelman, 1978; Mountcastle, 1979; Sporns et al., 2005; Watts and Strogatz, 1998), whose structural and functional connectivity is continuously shaped by the experience and the surrounding environment (Bressler and Tognoli, 2006). This constant environmental, experience-dependent modulation of neuronal connectivity is possible owing to synaptic plasticity, that is, the potential of neural activity to induce changes in synaptic strength in response to internal and external modifications or lesions (Bliss and Lomo, 1973; Citri and Malenka, 2008; Schaefer et al., 2017).
Because the plastic changes shape the brain network organization (Stampanoni Bassi et al., 2019), impaired synaptic plasticity alters the synchrony of both local and distributed neuronal oscillations and could promote brain network dysfunctions (Uhlhaas and Singer, 2006, 2010) in many neurological diseases, including movement disorders.
Movement disorders encompass a variety of complex conditions affecting the nervous system at multiple levels. The underlined pathological neurodegenerative processes alter the neuronal phasic and/or tonic firing discharge (Calabresi et al., 2007a, 2007b) leading to aberrant synaptic plasticity and, consequently, to alterations in functioning of large-scale brain networks (Eidelberg, 2009; Poston and Eidelberg, 2012). These abnormalities represent the functional correlates of the clinical patterns observed in movement disorders (Kreitzer and Malenka, 2008; Mink, 2003; Pisani et al., 2005). Therefore, the clinical phenomenology of these conditions is not limited to mere motor symptoms but entails also cognitive and motivational aspects resulting in the disruption of motor learning and motor behavior. Because of the neurodegenerative aspects, the responsiveness to pharmacological and surgical therapies, particularly as the disease progresses, is not uncommonly disappointing. In the latest years, the value of integrated rehabilitative programs for movement disorders has been demonstrated (Abbruzzese et al., 2016; Bloem et al., 2015; Ferrazzoli et al., 2018a, 2018b; Hohler et al., 2012; Mak et al., 2017; Ortelli et al., 2018). The effectiveness of rehabilitation probably relies on its potential to promote functional recovery acting as a modulator at both molecular (synaptic) level (Fontanesi et al., 2015; Hirsch et al., 2018; Sacheli et al., 2019) and circuitry level (Ahlskog, 2018; Voss et al., 2010). To foster adaptive processes, prevent maladaptive ones, and promote changes in neural circuitries, noninvasive brain stimulation techniques (NIBS), such transcranial magnetic stimulation (TMS) and transcranial direct current stimulation (tDCS), may serve as a complementary therapeutic modalities in clinical neurorehabilitation (Bashir et al., 2010; Hummel and Cohen, 2006; Liew et al., 2014; Page et al., 2015; Rroji et al., 2015; Schulz et al., 2013; Ziemann, 2017). Nonetheless, several gaps remain in our current understanding of the relationships among plasticity and neural networks, and how neurorehabilitation could promote clinical gains is still unclear. Deepening the knowledge in these fields is necessary for treating movement disorders. In this narrative review, we will describe first the main plasticity mechanisms and the neural networks underlying motor learning and motor behavior. Therefore, it will be outlined how the neuropathological alterations in movement disorders could provoke aberrant plastic changes, thus leading to these networks' disruption. On these bases, the potential contribution of neurorehabilitation in restoring the neural function will be discussed.
Motor Learning and Motor Behavior: Description of Plasticity Mechanisms
Long-term potentiation (LTP) and the converse process of long-term depression (LTD) are the most widely recognized physiological models of plasticity (Bliss and Gardner-Medwin, 1973). Both LTP and LTD may be induced by persistent stimulation of synapses (Bliss and Lomo, 1973; Calabresi et al., 1992; Clark, 1950; Nabavi et al., 2014) and may serve as the substrate for memory, learning, and behavior (Bailey and Kandel, 1993). LTP induces a persistent strengthening of synapses based on recent patterns of activity. The opposite LTD produces a long-lasting decrease in synaptic strength. Stimulation frequency is only one of the determinants of synaptic plasticity, which can also be induced by repetitive paired-pulse stimulation to presynaptic and postsynaptic neurons, depending on the spike timing: the so-called “spike timing-dependent plasticity” (STDP). STDP changes synaptic strength as a function of the timing between the presynaptic and the postsynaptic action potential (Bi and Poo, 2001; Markram, 1997). Therefore, changes in spike time of only few milliseconds can determine opposite effects on the synaptic strength (Markram, 1997).
Several signal transduction pathways, triggered by activation of both ionotropic and metabotropic receptor, generate LTP and LTD. N-methyl-D-aspartate (NMDA) receptor activation increases intracellular calcium concentration and represents the first step for the signaling cascades leading to cell depolarization and LTP. Noteworthy, elevated intracellular calcium concentration following NMDA receptor activation can also trigger LTD (Bear and Abraham, 1996). The inhibition mediated by gamma-aminobutyric acid (GABA) activity shapes the level of glutamate-induced depolarization. Similarly, excitatory inputs onto GABAergic interneurons regulate the likelihood of spiking in GABAergic cells and provide feedback or feedforward inhibition onto excitatory cells (Buzsáki and Eidelberg, 1982). These electrical signals are translated into molecular changes that modify the synaptic junctions allowing modifications in the neural connections (Kandel, 2001; Rioult-Pedotti, 2000; Sanes and Donoghue, 2000; Tibbetts, 2010). This model has been used to explain the long-term memory storage, and it is currently predominant in neuroscience. Nonetheless, there are theoretical arguments and experimental data against the vision of LTP and LTD as the sole mechanisms responsible for learning and memory (Abraham et al., 2019). An alternative posed to the synaptic plasticity model is the concept of the cell-intrinsic memory storage mediated by thermodynamically stable molecules (Johansson et al., 2014). Studies in mammals and invertebrates documented roles for DNA methylation in the formation of a variety of forms of learning and memory (Day and Sweatt, 2010). Other authors stated that cell bodies of neurons express molecular, nonsynaptic connections whereby the neurons can directly exchange information regarding learning-related interactions with each other (Landry et al., 2013). Noncoding RNAs, which are known to mediate neuronal learning-related epigenetic changes, could be exchanged via exosomes or tunneling nanotubes and might modulate the communication of information regarding the neural activity and the neural state (Abraham et al., 2019; Shepherd and Bear, 2011). Conceivably, both synaptic-specific plasticity and cell-wide intrinsic plasticity play critical roles in memory and learning (Langille and Brown, 2018).
Motor Learning and Motor Behavior: A Network Perspective
Motor learning and motor behavior result from the interaction among different and widespread brain areas that communicate with each other through neural pathways organized into a sort of “orthogonal system” (Ferrazzoli et al., 2018a). There are two principal sets of connections that control the different phases of motor learning into the networks: the first one proceeds in “horizontal direction,” by connecting the prefrontal, the sensorimotor, and the parietal cortices. The other one proceeds in “vertical direction,” by connecting the same cortical areas with the basal ganglia (BG), the cerebellum, and the spinal cord. The coordinated functioning of these areas allows to learn new skills and controls the motor performances during the action execution (Ferrazzoli et al., 2018a). Distinct alterations of networks' nodes and edges lead to the disruption of the motor, cognitive, and emotional aspects underlying the movement expression. Therefore, movement disorders cannot be regarded as mere “motor” pathologies, but rather as complex motor behavior diseases.
Motor learning, including the acquisition, consolidation, automatization, and retention of motor skill memories, implicates continuous functional and structural activity-dependent changes (Ganguly and Poo, 2013) at multiple levels (cortex, cerebellum, BG, amygdala, spinal cord) (Baraduc et al., 2004; Hübener and Bonhoeffer, 2010; Johansen et al., 2014; Li Voti et al., 2011; Muellbacher et al., 2002; Wolpaw and Tennissen, 2001; Yin et al., 2009). The process of learning is a multifaceted experience-dependent skill acquisition process based on implicit and explicit mechanisms for ensuring high-quality motor performances in any context. The main modalities of learning by which the skill acquisition process could be accomplished are represented by the unsupervised learning, the supervised learning, and the reinforcement learning (Doya, 1999, 2000, 2002). The unsupervised learning is a classical conditioning, whereby two or more stimuli produce an association to generate a mapping of the statistical regularities of the perceived environment (Hinton and Sejnowski, 1999). This is a slow form of learning that takes place in the cerebral cortical areas (Doya, 1999, 2000). The supervised learning is a shaping form of learning that provides a continuous comparison between the expected plan of action and what has been effectively realized for minimizing errors and reaching the desired goal. Supervised learning is fast and takes place in the cerebellum (Doya, 2000). Finally, the reinforcement learning is an operant conditioning based on reward prediction error: this is a slow form of learning, proceeding for “trial-and-error,” taking place in the BG (Doya, 2000).
The learning process goes through three stages (“cognitive,” “associative,” and “autonomous”) during which the cognitive system, the motivational system, and the motor system work together for acquiring skills and refining actions (Fitts and Posner, 1967). In the cognitive stage, the motor sequence to be learned is consciously planned in any single component and the movements are slow and often ineffective. During the associative stage, environmental cues and feedback are used to improve the action performance making the movements even more accurate, fluid, and efficient. Proceeding toward the final, autonomous stage, the motor sequence is honed into an automatized routine requiring very low cognitive demands. Dopamine (DA) plays a crucial role in these learning dynamics (Berridge, 2006; Schultz, 1998, 2006; Wise, 2004) modulating the networks that process both motivational stimuli and goal-directed behaviors. Activity in the D1-expressing, direct, striatonigral “go” pathway increases excitation of cortical activity and facilitates movement. By contrast, activity in the D2-expressing, indirect, striatopallidal “no-go” pathway increases inhibition of cortical activity and inhibits movement (Kravitz et al., 2010). Specifically, neurons in the “direct” pathway project directly from the putamen to globus pallidus pars interna (GPi)/substantia nigra pars reticulata (SNr), whereas striatal neurons in the “indirect” pathway connect the putamen with the GPi/SNr via synaptic connections in the globus pallidus pars externa (GPe) and subthalamic nucleus (STN). Activation of DA receptors on striatal medium spiny neurons (MSNs) modulates gating of ion channels altering the intrinsic excitability of these neurons (Surmeier et al., 2007): activation of D1 receptors increases the excitability of MSNs in the direct pathway, whereas activation of D2 receptors decreases the excitability of MSNs in the indirect pathway. Thus, the output activity of the BG is influenced by opposing effects of inhibitory inputs from the “direct” pathway and excitatory inputs from the “indirect” pathway. This model predicts that reduced activation of DA receptors, caused by DA deficiency, results in reduced inhibition of neurons in the indirect pathway and decreased excitation of neurons in the direct pathway (Obeso et al., 2000a). The success in predicting the effect of lesioning STN and GPi has reinforced this vision. However, several features are at odds with this concept. First of all, the model excludes evidence of dopaminergic innervation of extrastriatal regions and the role of striatal cholinergic interneurons (Obeso et al., 2000b). Furthermore, the functional anatomy of the motor circuitry of the BG is not only arranged in separate parallel circuits (Gerfen et al., 1990), but into a series of parallel and somatotopically segregated, highly collateralized projections that are regulated by several internal horizontal circuits (Joel and Weiner, 1997). Therefore, the motor circuits of the BG should be considered as a complex network formed by finely arranged cortico-BG-cortical parallel loops, which provide positive feedforward signaling for movement preparation and execution, and internal circuits, which mainly serve a feedback stabilizing function (Obeso and Lanciego, 2011; Obeso et al., 2000b). Following this idea, activation of cortical motor areas provides rapid and powerful disynaptic effects on the GPi/SNr that are either inhibitory (through the direct pathway) or excitatory (through the STN). These projections are suited for the control of repetitive movements and learned motor sequences. By contrast, the polysynaptic indirect pathway, which also has an excitatory effect on GPi/SNr, is more likely to be involved in motor learning processes (Obeso et al., 2000b).
Phasic responses of dopaminergic neurons are activated in the presence of reward-related stimuli and in relation to the discrepancy between the expected reward and what is really experienced (Schultz, 1998), ensuring adaptation to environmental changes (Horvitz, 2000; Mirolli et al., 2013). This dynamic, highly integrated learning system is implicated in the storage of memories (Caligiore et al., 2019) and allows to acquire and modulate the motor behaviors over time, space and environment. Other neuromodulators other than DA are involved in these learning processes: serotonin is important to drive reinforcement learning (Fischer and Ullsperger, 2017) and to regulate the interplay between supervised and reinforcement learning (Schweighofer et al., 2004). Noradrenaline regulates some components of “trial-and-error” learning (Doya, 2002), whereas acetylcholine consolidates the interaction between unsupervised and reinforcement learning (Hasselmo et al., 1995; Hasselmo, 2006).
As previously outlined, an orthogonal system of areas and connections subtends the dynamics of learning (Caligiore et al., 2019; Ferrazzoli et al., 2018a; Hikosaka et al., 2002). The networks co-work to elaborate input information, organize output signals, and generate the actions to be performed (Fig. 1). Striatum filters and synthesizes motor, cognitive, and emotional information coming from the cortex through processes controlled by the reciprocal dopaminergic/cholinergic innervation (Do et al., 2012). The external segment of globus pallidus is a key node in the control of the information flowing through the BG, whereas the internal segment integrates converging signals from cortical areas (via the striatum) (Saga et al., 2017). Movements or sequences that lead to good outcomes, based on the reward prediction error, are “reinforced” and finally “stored” in the putamen as “habits”: every motor schema, from the simplest movement of a single joint to the most complex motor sequence, is acquired through this learning process (Yelnik, 2008).
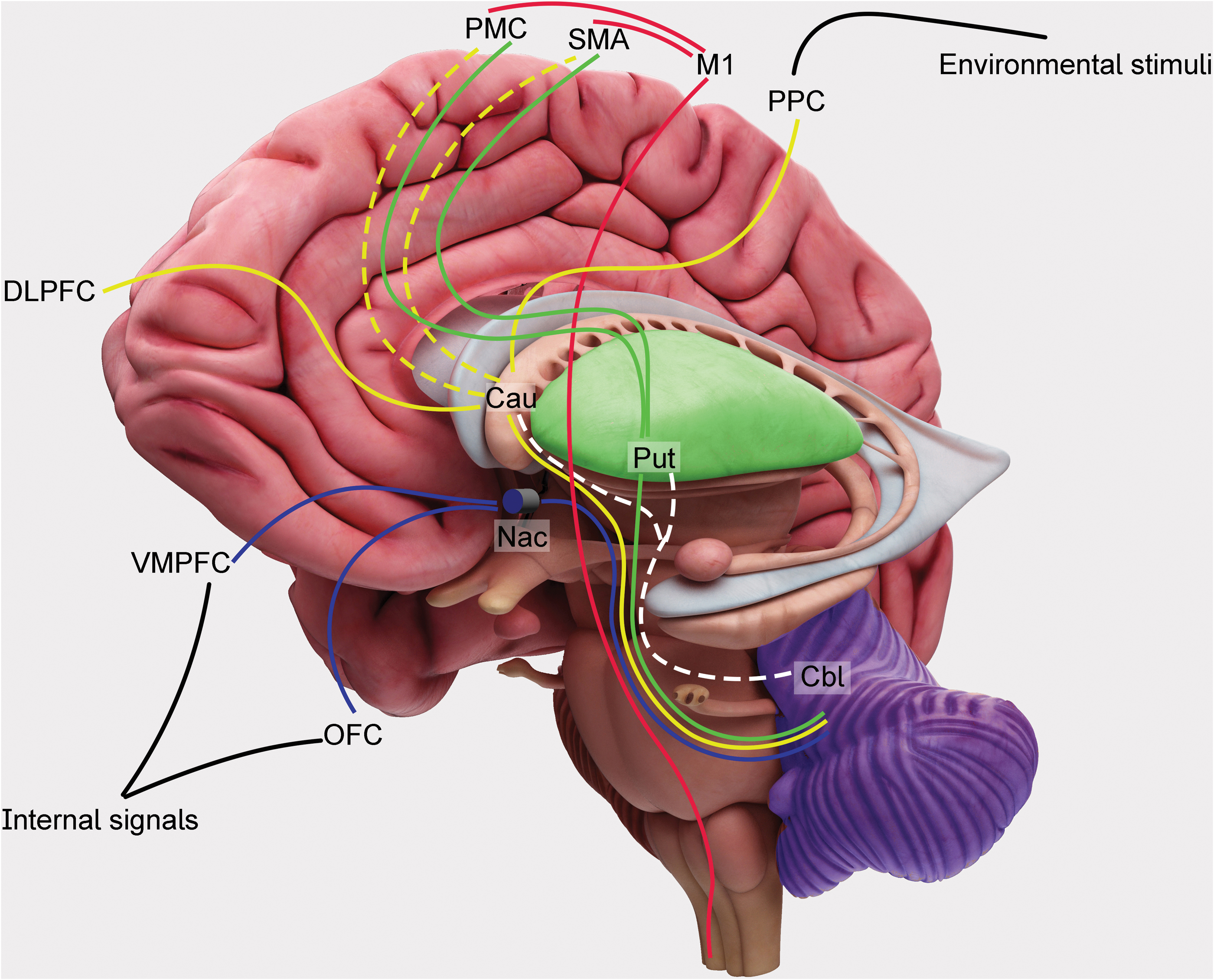
Hierarchical orthogonal system for motor learning and motor behavior. Internal signals (proprioceptive, vegetative, and visceral information) and sensorial inputs (arising from the environment) represent the bases of the hierarchical orthogonal system (see the text). The system intercepts the needs to be met, the goals to be achieved, and plans the actions to be performed. Subcortical areas, such as the hypothalamus and amygdala, after receiving the internal signals, activate the OFC, the VMPFC, and the nucleus accumbens (“Nac” in the figure), simultaneously. This network is classically known as limbic system. The cortical areas (such as visual and auditory cortices), after receiving sensory information, send it to the DLPFC and the PPC, to be integrated. This cortical elaboration is sent to the nucleus caudatum (“Cau” in the figure) to be filtered and elaborated. Therefore, it is sent back to PMC, SMA, and finally to M1 for the movement expression. Habitual motor schemata are selected from the putamen (“Put” in the figure) where the acquired skills and behaviors are stored. Cerebellum (“Cbl” in the figure) shapes and corrects the “ongoing” motor behaviors, typically modulating the striatal function through the deep nuclei and the thalamic relay. Continuous lines represent descending projections; dotted lines represent ascending projections. Cau, caudatum; Cbl, cerebellum; DLPFC, dorsolateral prefrontal cortex; M1, primary motor cortex; Nac, nucleus accumbens; OFC, orbitofrontal cortex; PMC, premotor cortex; PPC, posterior parietal cortex; SMA, supplementary motor area; VMPFC, ventromedial prefrontal cortex. Color images are available online.
Once an action has been acquired, the cerebellum ensures that its correct performance is maintained in the presence of changes in the environment: this adaptive process is called “motor adaptation” (Krakauer et al., 2019). Specifically, the cerebellum continuously evaluates the timing (Hikosaka et al., 2002) of the sequence to be performed and corrects errors of the “on-going” movements (Penhune and Steele, 2012). Other cortical and subcortical areas subtend the movement selection, the action planning, and the motor control. The pathway connecting the anterior insular cortex with the dorsal anterior cingulate cortex monitors external inputs and internal outputs basing on information saliency and controls whether other large-scale brain networks have to be activated or not (Bressler and Menon, 2010). The fibers connecting the dorsolateral prefrontal cortex (DLPFC), the posterior parietal cortex (PPC), and the inferotemporal cortex are part of the central executive network that is engaged in higher order cognitive demands and attentive control. These areas project toward the orbitofrontal cortex (OFC), the ventromedial prefrontal cortex (VMPFC), the limbic structures, and the caudatum for selecting which action has to be planned in response to the internal needs and to the external environmental context. The selected motor program is finally sent to the supplementary motor area (SMA) and to the premotor cortex (PMC) that project the information toward the primary motor cortex (M1) for movement execution (Caligiore et al., 2019).
The complex dynamics of these brain networks explains why movement disorders may result either from single node dysfunction or from aberrant communication among multiple nodes (Jinnah and Hess, 2006; Quartarone and Hallett, 2013; Schirinzi et al., 2016).
It will be now summarized how aberrant plastic changes shape the brain networks into a negative state of functioning in different movement disorders.
Aberrant Plasticity and Network Dysfunctions in Movement Disorders
Parkinson's disease
Parkinson's disease (PD) is characterized by motor, cognitive, and motivational abnormalities, which result in disorders in both movement expression and action performing (Mazzoni et al., 2012). The dopaminergic neuronal loss in substantia nigra pars compacta is universally regarded as the pathological hallmark of PD (Lees et al., 2009). The cornerstone of pharmacological treatment for PD is represented by DA replacement therapy (DRT), and
The dopaminergic innervation of the striatum is critical for higher order brain functions, including behavioral learning (Bergman et al., 1998; Centonze et al., 2001; Pisani et al., 2001). Converging sensorimotor, cognitive, and emotional information arriving from cortical pathways to the striatum generates a “compressed” and highly integrated output message to the frontal cortex, where the selection of a proper motor behavior is finally elaborated (Groenewegen, 2003; Leisman et al., 2014). The activation of DA receptors plays a key role in the induction of LTP and LTD, at both cortical and striatal level (Centonze et al., 2001). It follows that both the defective mesostriatal dopaminergic transmission and alterations in other neural pathways are responsible for abnormal corticostriatal plasticity, which finally subtends motor symptoms and cognitive dysfunction in PD (Bohnen and Albin, 2011; Calabresi et al., 2007a, 2007b; Picconi et al., 2005). Direct measurement of transmitter content during high-frequency stimulation of corticostriatal fibers showed that DA increases dramatically during the induction of synaptic plasticity (Calabresi et al., 1995). In normal conditions, the endogenous dopaminergic tone exerts a negative modulation on glutamatergic neurotransmission (Pisani et al., 2005). Neurophysiological, in vitro studies showed that after nigral lesion following unilateral 6-hydroxydopamine (6-OHDA) injections, striatal spiny neurons exhibit a prominent enhancement of glutamate-mediated activity (Calabresi et al., 1993). These finding suggest that DA denervation leads to increased glutamatergic cortical inputs and augments neuronal excitability in the striatum. Moreover, DA denervation induces supersensitivity of D2 receptors, reduces the inhibitory influence of D1 receptors, and prevents corticostriatal LTP (Calabresi et al., 1993; Centonze et al., 1999), as tested by TMS paradigms (Kishore et al., 2011). This defective striatal signaling is responsible for abnormal oscillatory activities in many areas of the orthogonal network. In 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated monkeys, it has been found that nigral degeneration is accompanied by loss of Purkinje cells and correlates with persistent activity in the cerebellum (Heman et al., 2012). Both normal striatal dopaminergic signaling and cerebellar sensory processing functions influence M1 plasticity. Thus, hyperactivation of the cerebellum in PD patients would prevent any discrete excitatory input from being efficiently processed and interferes with the cerebellar tuning of M1 plasticity (Kishore et al., 2014a, 2014b). This wide network disruption is reasonably involved in the entire clinical phenomenology observed in PD: (1) delayed initiation and execution of movements (Bergman et al., 1998), (2) aberrant expression of habitual, automatic, goal-based actions (Gilat et al., 2017; Redgrave et al., 2010; Wu et al., 2015a, 2015b), (3) dysregulation of reward-based (Schott et al., 2007) and error-based learning processes (Shiner et al., 2012), (4) impairment of switching from automatic to voluntary and goal-based actions (Hikosaka and Isoda, 2010; Kim and Hikosaka, 2015), and (5) impairment of motor planning (Goldenberg et al., 1986).
In the parkinsonian state,
Atypical parkinsonian syndromes
Atypical parkinsonian syndromes (APS) are a group of hypokinetic neurodegenerative diseases other than PD. Progressive supranuclear palsy (PSP), multiple system atrophy (MSA), and corticobasal degeneration (CBD) are the most prominent representatives of this group (Brooks, 2002).
The neuropathological hallmark of PSP is the hyperphosphorylated tau protein deposition, which is responsible for the formation of fibrillary aggregates (globose neurofibrillary tangles) in neurons and glia of numerous cerebral areas (Daniel et al., 1995) and for the significant loss of cortical interneurons in the SMA, M1, and thalamus (Halliday et al., 2005). Patients with PSP have abnormal M1 LTP/LTD-like plasticity (Conte et al., 2012): TMS studies of the motor cortex with theta burst stimulation have shown that patients with PSP exhibit greater facilitation than healthy subjects following LTP-like inducing protocols and paradoxical facilitation following LTD-like protocols (Conte et al., 2012). Interestingly, the enhanced LTP-like cortical synaptic plasticity parallels the disease progression (Conte et al., 2012). These data suggest that PSP represents a network-based brain disorder. Gardner and colleagues (2013) mapped intrinsic connectivity to the dorsal midbrain tegmentum (dMT) in healthy controls and in patients with PSP. While healthy controls showed a convergent pattern of connectivity to the dMT (including brainstem, cerebellar, diencephalic, BG, and cortical regions), the authors found significant connectivity disruptions within this network in patients with PSP. Notably, patients with more severe functional impairments showed lower mean dMT network connectivity scores (Gardner et al., 2013).
The principal neuropathological characteristic of MSA is the presence of aggregates containing argyrophilic neuronal and glial cytoplasmic inclusions of the synaptic protein α-synuclein (Grazia Spillantini et al., 1998) mainly in the striatum, substantia nigra, brainstem nuclei, dentate nuclei of the cerebellum, intermediolateral columns of the spinal cord, and Onuf's spinal nucleus (Brooks, 2002). The strength of structural connectivity among BG, ventral diencephalon, thalamus, and cerebellum (measured as the number of streamlines derived from tractography) is reduced in MSA patients compared with PD patients and healthy controls (Abos et al., 2019). α-Synuclein is involved in synaptic plasticity (Cheng et al., 2011), and its alteration in MSA could be responsible for abnormal plastic changes. Coherently, TMS studies with theta burst stimulation have revealed a lack of response to both LTP and LTD plasticity-inducing protocols (Suppa et al., 2014).
The main neuropathological feature of CBD consists of asymmetrical degeneration of posterior frontal cortex, inferior parietal cortex, superior temporal cortex, thalamus, substantia nigra, and cerebellar dentate nuclei. Neuronal and glial pathological lesions containing hyperphosphorylated microtubule-associated tau protein (Pick cells) are specific of the disease (Brooks, 2002). TMS testing of the affected hemisphere showed abnormal plasticity with a great intersubject variability (Suppa et al., 2016). Some authors found asymmetric M1 excitability, reduced intracortical and interhemispheric inhibition, and abnormal integration of somatosensory afferent inputs in the motor and sensory cortices (Di Stasio et al., 2019), suggesting that, as is the case with many other neurodegenerative diseases, CBD appears to be a neural network disorder.
Dystonia
Dystonia can be defined as hyperkinetic movement disorder characterized by prolonged muscle contractions, which causes involuntary repetitive twisting movements and abnormal postures of the affected body parts (Fahn et al., 1998). Dystonia shares several molecular and synaptic abnormalities with other conditions, such as
TMS studies have shown increased LTP-like plasticity suggesting disruption of homeostatic regulation (Quartarone and Hallett, 2013; Quartarone and Pisani, 2011). Therefore, the positive-feedback nature of LTP may trigger an uncontrolled increase in synaptic effectiveness, which can foster a form of maladaptive plasticity and destabilize the neural networks (Quartarone and Hallett, 2013; Quartarone and Pisani, 2011; Quartarone et al., 2006). This is coherent with the clinical observation that some forms of focal dystonia are triggered by periods of intensive, task-specific training (Quartarone et al., 2006). In these cases, aberrant associations between sensory inputs and motor outputs, due to a deficient synaptic scaling, lead to pathological representations of specific motor schemata (Quartarone and Pisani, 2011). It follows that task-specific dystonias could rise from certain circuits breakdown responsible in turn for the consolidation of abnormal neural engrams containing redundant information (Quartarone and Hallett, 2013; Quartarone and Pisani, 2011). Silberstein (2003) tested the hypothesis that there are distinct temporal patterns of synchronized neuronal activity in the pallidum that characterize patients with dystonia and PD. The authors recorded local field potentials (LFPs) from the caudal and rostral contact pairs of macroelectrodes implanted into the pallidum. In patients with dystonia, percentage LFP power was less in the 11–30 Hz band and greater in the 4–10 Hz band compared with untreated or treated PD patients.
Abnormal oscillatory activities and aberrant synchrony among BG, thalamus, and cortex disrupt the normal sensorimotor processing required for movement expression in dystonia (Liu et al., 2002, 2008; Quartarone and Hallett, 2013; Silberstein, 2003). Cerebellar abnormalities may also play a causative role (Prudente et al., 2014), due to the involvement of the cerebellum in temporal, spatial, sensorimotor discrimination (Restuccia, 2001). Taken together, these data support the concept of dystonia as network disorder (Schirinzi et al., 2018).
Huntington disease
HD is a progressive neurodegenerative disorder inherited in an autosomal dominant manner caused by expansion of a polymorphic CAG triplet repeat in exon 1 of the huntingtin (HTT) gene. Patients exhibit motor deficits, cognitive disorganization, and loss of emotional control (Bates et al., 2015; Naarding et al., 2001).
CAG encodes the amino acid glutamine, and the expansion gives rise to proteins presenting abnormally long polyglutamine regions, which form aggregates thought to interfere with vital cell functions. The brain structures most severely affected by HD are the striatum and its anatomical and functional connections with the cerebral cortex. Substantial reduction in striatal size, cortical atrophy (mainly in the deep layers III, V, VI), and astrogliosis has been described as striking morphological features in HD (Lobsiger and Cleveland, 2007; Vonsattel and DiFiglia, 1998). Interestingly, long before cell loss occurs, HD impairs the mechanisms by which cortical and striatal neurons communicate (Rebec, 2018). Indeed, recordings of spike and LFP activity of mice behaving mHTT indicate that the close relationship between cortical output neurons and striatal MSNs is disrupted (Rebec, 2018). Moreover, it has been found that silencing the normal HTT gene in developing mouse cortex leads to excessive excitatory synapses formation and, therefore, abnormal corticostriatal connectivity (McKinstry et al., 2014). All together, these data indicate that normal HTT is required for establishing physiological connectivity at corticostriatal level. Consistent with this view, LTP studies from HD mouse models have demonstrated the presence of synaptic abnormalities at striatal synapses and alterations in long-term plasticity (Cepeda et al., 2003). Different observations strongly suggest that these abnormalities are subtended by deficits in the glutamatergic signaling/transmission/gene expression (Centonze et al., 2006; Cepeda et al., 2001, 2003), although other neurotransmitters have been implicated (Rebec, 2018; Smith-Dijak et al., 2019). Not surprisingly, studies in genetic and phenotypic animal models of HD have shown the presence of LTP alterations (Picconi et al., 2006). The increased excitability in striatal MSNs and the following loss of flexibility in striatal neural processing lead to large-scale corticostriatal abnormalities and to alterations of the motor–behavioral-related dynamics in HD (Hong et al., 2012; Raymond et al., 2011; Rebec, 2018). Crupi and colleagues (2007) tested cortical and brainstem LTP-like plasticity in 8 symptomatic HD patients and 10 healthy controls. The authors demonstrated impairment of both cortical and brainstem LTP-like plasticity in HD patients, which was properly similar to LTP deficits previously reported in HD animal models (Crupi et al., 2007).
Cerebellar ataxias
The definition of cerebellar ataxia refers to a series of condition whose origin can be traced back to deficits in the cerebellum and/or cerebellar pathways, which play a role in motor learning, coordination, and fine movements adjustment. As previously stated, the cerebellum is part of the wide-distributed learning network and plays a pivotal role in error-based motor learning and in motor adaptation (Doyon and Benali, 2005; Hikosaka et al., 2002; Marsden and Harris, 2011). LTP and LTD at cerebellar parallel fiber-Purkinje cell synapses have long been seen as critical for motor control and motor learning (Jörntell and Hansel, 2006; Rinaldo and Hansel, 2010). It follows that adjustments of movements and motor coordination provided by the cerebellum require both intact LTD and LTP (Rinaldo and Hansel, 2010). Some forms of ataxia are caused by mutations in genes encoding for ion channels that are relevant for the induction of synaptic plasticity (Rinaldo and Hansel, 2010). Spinocerebellar ataxia type 6 (SCA6) and episodic ataxia type 2 (EA2) result from mutations in CACNA1A gene encoding for the alpha-1a subunit of the P/Q-type voltage-gated calcium channel (Ophoff et al., 1996). These channels, abundantly expressed in Purkinje cells, are involved in transmitter release at synaptic terminals. Hoebeek and colleagues (2005) found that mice suffering from a spontaneous point mutation in the CACNA1A gene show a disruption in the firing regularity of Purkinje cells and exhibit gross motor deficits similar to those seen in humans suffering from SCA6 and EA2. Therefore, aberrant synaptic plasticity could play a role in the pathogenesis of cerebellar ataxias (Rinaldo and Hansel, 2010). Indeed, few studies have shown that patients with cerebellar ataxia display lack of LTP-like effect induced by both sensorimotor and cerebellar-motor associative stimulation protocols (Dubbioso et al., 2015; Lu et al., 2017). Interestingly, applying a connectivity-based approach, Tzvi and colleagues (2017) found that patients with cerebellar degeneration present increased activity in parahippocampal cortex and in cerebellar Crus I compared with healthy controls (Tzvi et al., 2017). Moreover, negative modulation of connections from SMA to M1 was present in ataxic patients with degeneration in cerebellar lobules IV-V-VI. The same subjects presented learning impairments (Tzvi et al., 2017). These findings suggest that the SMA-M1 connectivity is critically dependent on cerebellum (Arai et al., 2011; Fox et al., 1997; Tzvi et al., 2017) and that disruption of cerebellar synaptic plasticity could determine brain network dysfunctions and motor learning impairments in patients with cerebellar degeneration.
Gilles de la Tourette syndrome
Gilles de la Tourette syndrome (GTS) is a common neurodevelopmental disorder characterized by the persistence of motor and vocal tics often associated with psychiatric comorbidity leading to heterogeneous clinical expressions (Cavanna et al., 2009). Different studies demonstrated both decreased number of striatal GABAergic and cholinergic interneurons and abnormalities in pallidal projections (Kalanithi et al., 2005; Kataoka et al., 2010). These conditions result in global and fine-scale functional disorganization of cortico-BG networks in GTS (Worbe et al., 2012). Worbe and colleagues (2012) using resting-state functional MRI data showed stronger global integration and functional disorganization of cortico-BG circuits (loss of hubs, shorter path length, higher number of and stronger functional connections among the networks) in GTS patients compared with healthy controls. Also, in the case of GTS, abnormal plasticity could be the cause of this altered pattern of connectivity. Indeed, early studies using TMS and paired-pulse technique showed changes in M1 (shortened cortical silent period and reduced intracortical inhibition) and in brainstem excitability (significant increase in the mean duration of the R2 component of blink reflex) (Smith and Lees, 1989; Ziemann et al., 1997) that might represent the consequences of abnormal plasticity in GTS.
To test this hypothesis, Suppa and colleagues (2011) studied cortical plasticity and brainstem plasticity in 12 GTS patients, which were compared with 24 healthy controls. Cortical plasticity was tested by conditioning left M1 with intermittent or continuous theta burst stimulation in two separate sessions. Test stimulation consisted of 20 motor-evoked potentials (MEPs) recorded from first interosseus muscle before and after theta burst stimulation. Brainstem plasticity was tested by conditioning the right supraorbital nerve with facilitatory or inhibitory electric high-frequency stimulation (delivered at the same time as the late response of blink reflex or before the late response, respectively). Test stimulation consisted of 10 blink reflexes from the right orbicularis muscle before and after high-frequency stimulation. The authors found that none of the patients had the expected changes, neither in MEP amplitudes nor in the late component of the blink reflex (Suppa et al., 2011). The lack of these expected inhibitory and facilitatory changes confirms properly that abnormal plasticity in M1 and in brainstem plays a role in the neural network disruption in GTS (Suppa et al., 2011; Worbe et al., 2012). In another study in mild GTS patients (Brandt et al., 2014), the application of a paired associative stimulation (PAS) protocol (Stefan, 2000) failed to induce LTP-like synaptic plasticity but rather induced an unexpected LTD-like effect that inversely correlated with symptom severity (Brandt et al., 2014). In contrast, Martín-Rodríguez and colleagues (2015) reported abnormally increased LTP-like motor plasticity after a PAS protocol, however, in severely affected GTS patients.
Role of Rehabilitation in Modulating Aberrant Plasticity and Network Dysfunctions in Movement Disorders
Neurorehabilitation is based on the set of clinical and carer interventions aimed to recovery from a nervous system damage (due to acquired injuries or to degenerative diseases) for reducing or compensating the functional disturbances through the use of the patients' neuroplastic resources (Ferrazzoli et al., 2020). As previously outlined, alterations of synaptic plasticity promote brain network dysfunctions and subtend the onset and the progression of movement disorders (Fig. 2). Nevertheless, neuroplasticity is not lost even in the context of neurodegeneration. Plasticity is driven by behavioral, sensory, and cognitive modalities: it represents the main mechanism by which the damaged brain encodes experiences and relearns lost skills in response to rehabilitation (Ganguly and Poo, 2013; Kleim and Jones, 2008). Activity remains the main force for inducing neuroplasticity in the nervous system (Ganguly and Poo, 2013) and for setting the brain networks into new functional states (Ganguly and Poo, 2013). These activity-dependent changes allow patients with acute and chronic brain disorders (Bavelier et al., 2010) to gain motor, functional, and cognitive benefits (Cramer et al., 2011) in the short- and long-term (Mak et al., 2017; Wolf et al., 2006).
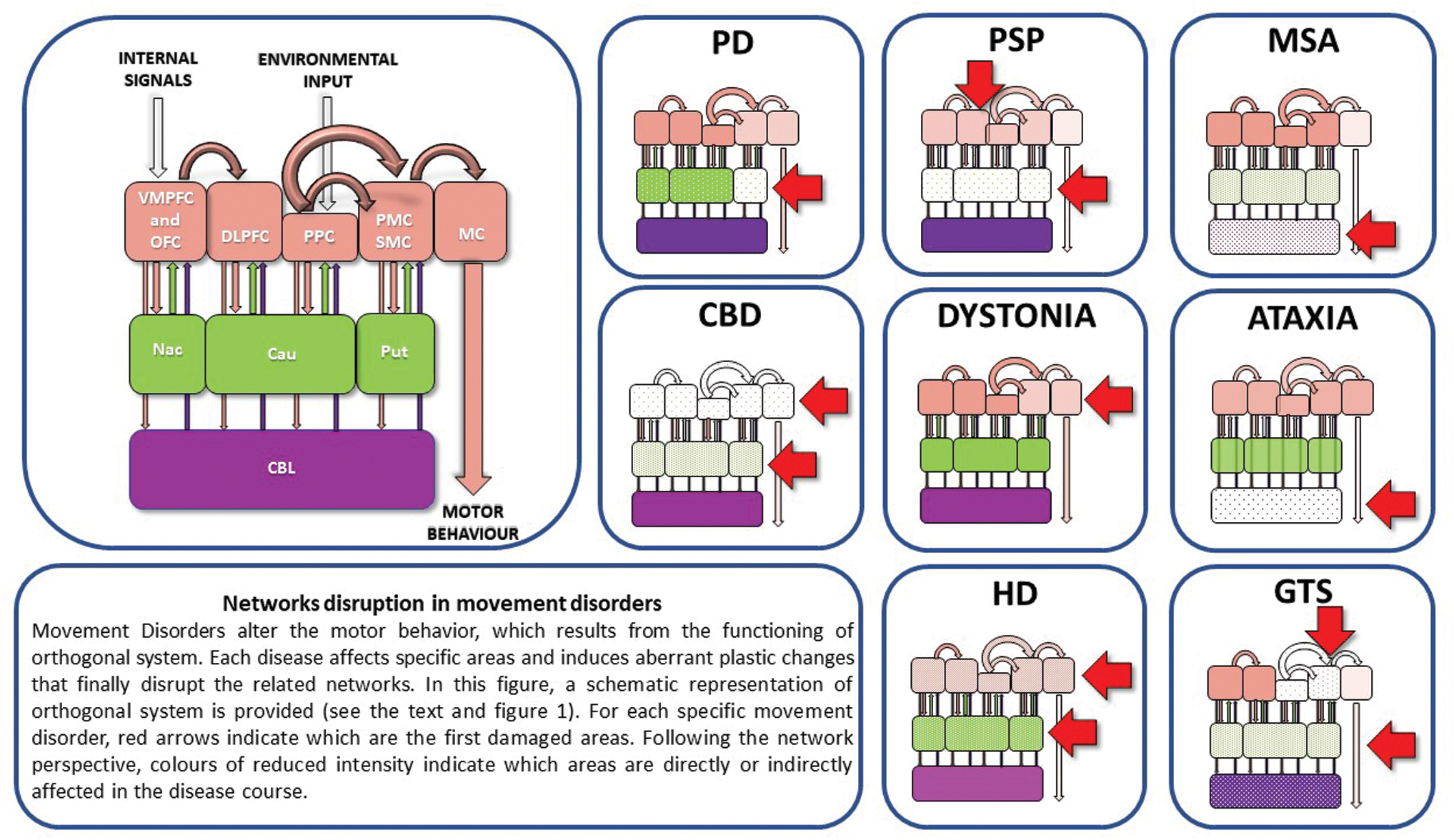
Network disruption in movement disorders. See the box. Cau, caudatum; CBD, corticobasal degeneration; CBL, cerebellum; GTS, Gilles de la Tourette syndrome; HD, Huntington's disease; M1, primary motor cortex; MSA, multiple system atrophy; Nac, nucleus accumbens; PD, Parkinson's disease; PSP, progressive supranuclear palsy; Put, putamen. Color images are available online.
It is known that experience-dependent changes can occur from the molecular and synaptic levels to large-scale neural network levels (Karni et al., 1998). Literature data show that only following certain modalities rehabilitation could drive adaptive changes in the networks spared from the disease and compensate for the damage/degeneration (Cheetham and Finnerty, 2007; Fox, 2009; Wang et al., 2016). Therefore, understanding what kind of rehabilitative programs have to be applied is crucial.
Driving plasticity to the right direction: cognitive-based training and aerobic exercise
Generally, cognitive-based skills acquisition training induces significant changes in the pattern of neural connectivity among widely distributed network nodes; in comparison, the mere physical activity leads to increases in connectivity predominantly in the motor cortex (Büchel, 1999; Kelly et al., 2006; Kleim et al., 1996; McIntosh et al., 1999; Monfils et al., 2016; Nudo, 2003). Therefore, because the training mode determines the activation of specific areas and pathways (Kleim and Jones, 2008), the major challenge in the field of movement disorders rehabilitation is finding the right modalities to act at both motor and cognitive circuitries and achieve clinical gains (Jakowec et al., 2016; Pascual-Leone et al., 2005). Converging evidence suggests that exercise has to be planned in terms of “learning modality” (Jakowec et al., 2016), that is, by merging cognitive, skill-based practice with aerobic exercise (Ferrazzoli et al., 2018a; Jakowec et al., 2016). Skill-based practice can be defined as a form of “goal-oriented” activity in which the performer has to achieve predetermined aims with temporal and spatial accuracy (Jakowec et al., 2016). Aerobic exercise is a form of training aimed to improve the oxygen consumption for enhancing the circulatory and the respiratory efficiency. Performing aerobic exercise has been related to neurotrophin signaling pathway activation (Phillips et al., 2014), angiogenesis (Van der Borght et al., 2009), neurogenesis (van Praag, 2005; van Praag et al., 1999), reduction in neuroinflammation (Real et al., 2017), and in oxidative stress (Roque et al., 2013). Studies from animal models showed that while aerobic exercise leads to increases in the density of blood capillaries, but no increase in the number of synapses, skilled-based practice generates greater number of synapses per neuron, but no increase in the capillary density (Garcia et al., 2012). Skilled-based practice results in broader functional connectivity in comparison with aerobic exercise but requires greater flexibility and effort in cortical processing (Jakowec et al., 2016). The modifications in connectivity arise from practice-related changes in synaptic plasticity at motor and nonmotor circuitries (Hund-Georgiadis and von Cramon, 1999; Kelly et al., 2006): this is associated with the creation of “internal” motor behavioral “models” based on the practiced tasks (Shadmehr, 1997). While cognitive, skill-based practice triggers connectivity, aerobic exercise is crucial to strengthen plasticity as it promotes the development of favorable biological and physiological environments in which the synaptic connectivity can take place (Jakowec et al., 2016). Indeed, many factors induced by aerobic exercise by priming the brain environment promote and consolidate synaptic neuroplasticity (Jakowec et al., 2016; Petzinger et al., 2013). Among these factors, there are neurotrophins, such as BDNF: the expression of this molecule increases in response to aerobic exercise in different regions of the brain (Cotman et al., 2007). The interaction between BDNF and its receptor TrkB can lower the threshold for the induction of LTP, both in healthy individuals and in those with neurological diseases (Yoshii and Constantine-Paton, 2010). In this concern, many studies (Petzinger et al., 2013) suggested that combining goal-based training with aerobic practice improves automaticity and cognitive motor control, thus reducing the attentional demands in complex behaviors, such as walking (Yogev et al., 2005).
Certain exercise parameters have to be set to induce plasticity: intensity, complexity of practice, and specificity. High-intensity interval exercise, rather than moderate-intensity continuous exercise, creates optimal conditions for inducing LTP-like synaptic plasticity (Andrews et al., 2019).
Moreover, according to a cross-sectional study (Alwardat et al., 2019), the cerebrospinal fluid Aβ42 content seems to improve in parallel with the increase of the total amount of physical activity in cognitively normal parkinsonian subjects, thus supporting the probable role of physical activity in decreasing the risk of dementia in PD.
Moreover, the exercise must be challenging and complex enough to stimulate learning from experience and foster the self-awareness. Finally, the task must be specific to engage the neural circuits affected by the disease and to drive the re-acquisition of lost skills (Jakowec et al., 2016).
In this concern, the direct and indirect effect of exercise and integrated motor–cognitive rehabilitation modalities have been described, both in animal models and in humans with movement disorders. Using the MPTP mouse model of PD, it has been found that daily treadmill exercise leads to (1) increased evoked DA release, (2) increased extracellular DA through downregulation of DA transporter expression, and (3) decreased clearance using fast-scan cyclic voltammetry within the dorsal striatum (Fisher et al., 2004; Petzinger et al., 2007). In the same model, using Western immunoblotting analysis of synaptoneurosomes and in vivo positron emission tomography (PET) imaging with DA-D2R-specific ligand [18F]fallypride, Vučcković and colleagues (2010) observed an increase in striatal DA-D2R expression in the striatum. In a translational study, Fisher and colleagues (2013) examined whether intensive exercise leads to similar alterations in DA-D2R expression using PET imaging with [18F]fallypride in individuals with early-stage PD: they found an increase in DA-D2R binding potential following an 8-week training program that was not observed in people who did not exercise. Moreover, people who exercised improved their postural control, suggesting that exercise can lead to neuroplasticity in dopaminergic signaling and contribute to improved function that may be task specific in early-stage PD (Fisher et al., 2013).
The neuroplasticity-based effects of exercise have been testified well beyond the modulation of dopaminergic signaling and transmission. By modifying the AMPA receptor subunit expression onto MSNs, intensive exercise leads to increased synaptic strength in the striatum (Kintz et al., 2013). Exercise may also impact on glutamate neurotransmission by reducing the presynaptic release of glutamate and modulating the intrinsic excitability properties of nigral DA neurons (Chen et al., 2017).
Moreover, there are several evidences linking exercise, plasticity, and improvements of motor functions with neurotrophic factors (Svensson et al., 2015). One study in mice treated with lipopolysaccharides (LPS) revealed that the beneficial effects of exercise are due to the activation of the BDNF-TrkB signaling pathway in neurons (Wu et al., 2011). Coherently with these findings, it has been found that treadmill-running restores LPS-impaired neurogenesis (Wu et al., 2007) and protects MTP-treated mice from motor deficits (Lau et al., 2011). Interestingly, these animals demonstrated also increased BDNF levels and improvement in mitochondrial function (Lau et al., 2011). Total BDNF mRNA is properly reduced in the hippocampus of HD mice, but interestingly, the wheel-running training increases total BDNF gene expression in these animals (Zajac et al., 2010). These data provide new insight into the role of BDNF in HD pathogenesis in addition to the mechanisms regulating normal BDNF gene expression (Zajac et al., 2010).
It is noteworthy that exercise paradigms leading to plastic changes may improve cognition other than motor aspects (Intzandt et al., 2018; Petzinger et al., 2013). Duchesne and colleagues (2016) found a functional reorganization of brain activity in cerebral regions concerned with motor learning (hippocampus, striatum, and cerebellum) in 19 early-stage PD patients following a 12-week progressive aerobic training. These functional changes were accompanied by improvements on behavioral outcomes. These findings explain the improvement in executive functions following aerobic exercise (Tabak et al., 2013) and suggest that physical activity could reorganize and/or activate the frontal brain networks. In this regard, Weng and colleagues (2017) found in a sample of healthy younger and older adults that 30 min of moderate-intensity aerobic cycling selectively increased synchrony among the brain regions associated with affect and reward processing, learning, and memory.
It is noteworthy that these findings are in agreement with a previous randomized controlled trial describing the effects of 24 weeks of structured exercise interventions in PD (David et al., 2015): this study provided Class IV level evidence for progressive resistance exercise or modified fitness counts in improving attention and working memory in nondemented PD patients with mild-to-moderate disease severity.
Beyond motor–cognitive rehabilitation: how NIBS can steer plasticity and modulate the activity of neural networks
All these data confirm that driving neural plasticity is likely to aid the treatment of a variety of brain diseases (Kilgard, 2012), including movement disorders. It is conceivable that neural plasticity might be targeted to reset dysfunctional circuits: in this perspective, neurorehabilitation could be considered as a valuable tool to manipulate fine-scale neural connectivity in human brain networks. Nevertheless, a lot of gaps in our knowledge of this process remain. An important limiting factor is to clearly understand the complexity of neural coding (Kilgard, 2012) and the exact role that exercise-induced plasticity plays in certain physiological and pathological conditions. This is the case of some forms of dystonia caused by maladaptive plasticity following intense, repetitive trainings (Quartarone and Hallett, 2013). Therefore, the efficacy of conventional rehabilitation treatments targeting neurological impairments could vary widely. NIBS can be applied over selected cortical and cerebellar regions to shape (facilitate or inhibit) their excitability, steer neural plasticity, modulate the activity of cortical–subcortical–cerebellar networks, and enhance retention of motor skills (Ganguly et al., 2020; Marchesi et al., 2019; Moisello et al., 2015; Page et al., 2015; Wu et al., 2008). In this concern, the application of repetitive TMS (rTMS) and tDCS in the treatment of movement disorders may be of particular interest (Benussi et al., 2020; Berardelli and Suppa, 2013; Brusa et al., 2005; Goodwill et al., 2017; Grados et al., 2018; Hamada et al., 2008; Liu et al., 2018; Manenti et al., 2015; Quartarone et al., 2017; Vanacore and Canevelli, 2019; Wu et al., 2008). High-frequency rTMS (>5 Hz) and anodal tDCS typically increase cortical excitability, whereas low-frequency rTMS (<1 Hz) and cathodal tDCS result in the opposite effect (Hallett, 2007; Medeiros et al., 2012).
rTMS at 10 or 0.5 Hz over M1 has been found to improve motor performance in “off-drug” patients with PD: high-frequency rTMS decreases rigidity and bradykinesia in the upper limb contralateral to the stimulation, whereas low-frequency rTMS reduces upper limb rigidity bilaterally and improve walking (Lefaucheur et al., 2004). Ikeguchi and colleagues (2003) found motor and functional improvements after frontal 0.2 Hz rTMS (six times for 2 weeks) and reduced regional cerebral blood flow in the cortical areas around the stimulated site. These findings suggest that the outlasting inhibitory effects of successive 0.2 Hz rTMS on the stimulated frontal cortical areas and the indirect effects on subcortical structures are related to improved parkinsonian symptoms (Ikeguchi et al., 2003).
In a double-blind placebo-controlled study (Hamada et al., 2008), it has been investigated whether modulation of SMA excitability with 5 Hz rTMS (once a week for the first 8 weeks) engenders therapeutic effects on motor symptoms in PD. The authors found significant improvements in total scores and motor scores of the Unified Parkinson's Disease Rating Scale.
In a double-blinded study, Fregni and colleagues (2006) aimed to study the effects of tDCS on motor function, simple reaction time (sRT), and corticospinal motor excitability in PD. The authors found that anodal stimulation of M1 significantly increased MEP amplitude and area and was associated with improvements of motor function and sRT. These results extend the notion that NIBS might improve motor function in patients with PD (Fregni et al., 2006).
Even in the case of APS, NIBS seem to provide such clinical gains (Kwon et al., 2011; Liu et al., 2018; Manenti et al., 2015; Manor et al., 2019; Mantovani et al., 2006; Shiga et al., 2002; Valero-Cabré et al., 2019; Vanacore and Canevelli, 2019). Anodal tDCS over the left DLPFC and cathodal tDCS over the right DLPFC provided 12 PSP patients with benefits in semantic and lexical skills in a sham-controlled, double-blind cross-over study (Valero-Cabré et al., 2019). Liu and colleagues (2018) found that 5-Hz rTMS targeting the cerebellum and bilateral M1 leads to increase in resting-state complexity within the motor network and improvements in motor control in MSA.
The extent to which anodal tDCS over the parietal cortex could facilitate naming performance has been analyzed in CBD subjects (Manenti et al., 2015). Manenti and colleagues (2015) demonstrated a shortening of naming latency and suggested that anodal tDCS over the left parietal cortex modulates the brain network implicated in action observation and representation (Manenti et al., 2015).
In dystonia, one possible strategy is an increase of inhibitory mechanisms. It has been reported that 30 min of inhibitory low-frequency 1-Hz rTMS stimulation over M1 may reduce writing pressure for at least 3 h in patients with focal hand dystonia (Siebner et al., 1999). Similarly, 1-Hz rTMS over PMC improves handwriting velocity and hand discomfort during writing (Tyvaert et al., 2006). Murase and colleagues (2005) compared the effect of subthreshold low-frequency (0.2 Hz) rTMS in three different motor areas including PMC in patients with focal hand dystonia. Stimulation of the PMC, none of the MC, significantly improved the rating of and prolonged the silent period. rTMS over the other sites revealed no physiological or clinical changes, suggesting that the lack of inhibition in the MC is secondary to the hyperactivity of PMC neurons. The somatosensory cortex, anterior cingulate cortex, and cerebellum represent other targets, which have been stimulated in dystonic patients, often with discrepant results (mainly due to the different parameters of stimulation) (Quartarone et al., 2017).
The use of NIBS has been also applied for the treatment of cerebellar ataxia (Benussi et al., 2020). In a double-blind sham-controlled trial, Shiga and colleagues (2002) found a significant alleviation of truncal ataxia in patients with spinocerebellar degeneration treated by active, 21-day TMS over both the cerebellar hemispheres and the inion. A recent pilot, randomized controlled trial (Manor et al., 2019) showed that a 4-week, 20-session rTMS intervention targeting the cerebellum leads to improvement in the scale for the assessment and rating of ataxia (SARA) scores and in objective metrics of postural sway during eyes-opened and eyes-closed standing.
In patients with HD, modulation of M1 excitability with rTMS might be therapeutically beneficial (Brusa et al., 2005). Groiss and colleagues (2012) analyzed the effects of M1-rTMS at different frequencies (1 and 10 Hz) on motor and cognitive functions in eight right-handed patients with HD in a blinded, sham-controlled cross-over study. Ten hertz rTMS shortened choice RTs when performed with the left hand and prolonged simple RTs when performed with the right hand. One hertz rTMS led to a sustained improvement in mood.
Clinical trials using both tDCS and rTMS have explored the efficacy of noninvasive brain stimulation also as a treatment for GTS (Grados et al., 2018). It has been found that 12-week, slow-frequency rTMS treatment over SMA improves overall tics in GTS patients (Kwon et al., 2011). Mantovani and colleagues (2006) tested whether low-frequency rTMS over SMA (10 daily sessions at 1 Hz, 100% of motor threshold, 1200 stimuli/day) could normalize overactive motor cortical regions and improve symptoms in subjects with obsessive-compulsive disorder or GTS. The authors found that symptoms improvement following rTMS was correlated with a significant increase of the right resting motor threshold and was stable at 3 months of follow-up.
Conclusions
Growing amount of data supports the converging evidence that many neurological and psychiatric diseases, including movement disorders, are linked to disruption of synaptic plasticity resulting in neural network dysfunctions. Often, the synaptic plastic changes precede the appearance of neuropathological alterations, thus raising the question of whether these are the cause or the consequence of the functional alterations. There are no definitive cures for movement disorders. Understanding the deep ties linking aberrant plasticity to neural network dysfunction will become increasingly urgent in the next future. Indeed, deepening the knowledge in this field will allow to implement rehabilitative programs targeting the underlying pathophysiology of movement disorders. To this end, the development of dedicated interventions harnessing plastic rearrangements that are capable to “drive” the “recovery” of neural networks physiology will be crucial. In this concern, merging aerobic training with goal-based practice, as well as the use of NIBS, appears as the master way to follow at this time (Fig. 3).
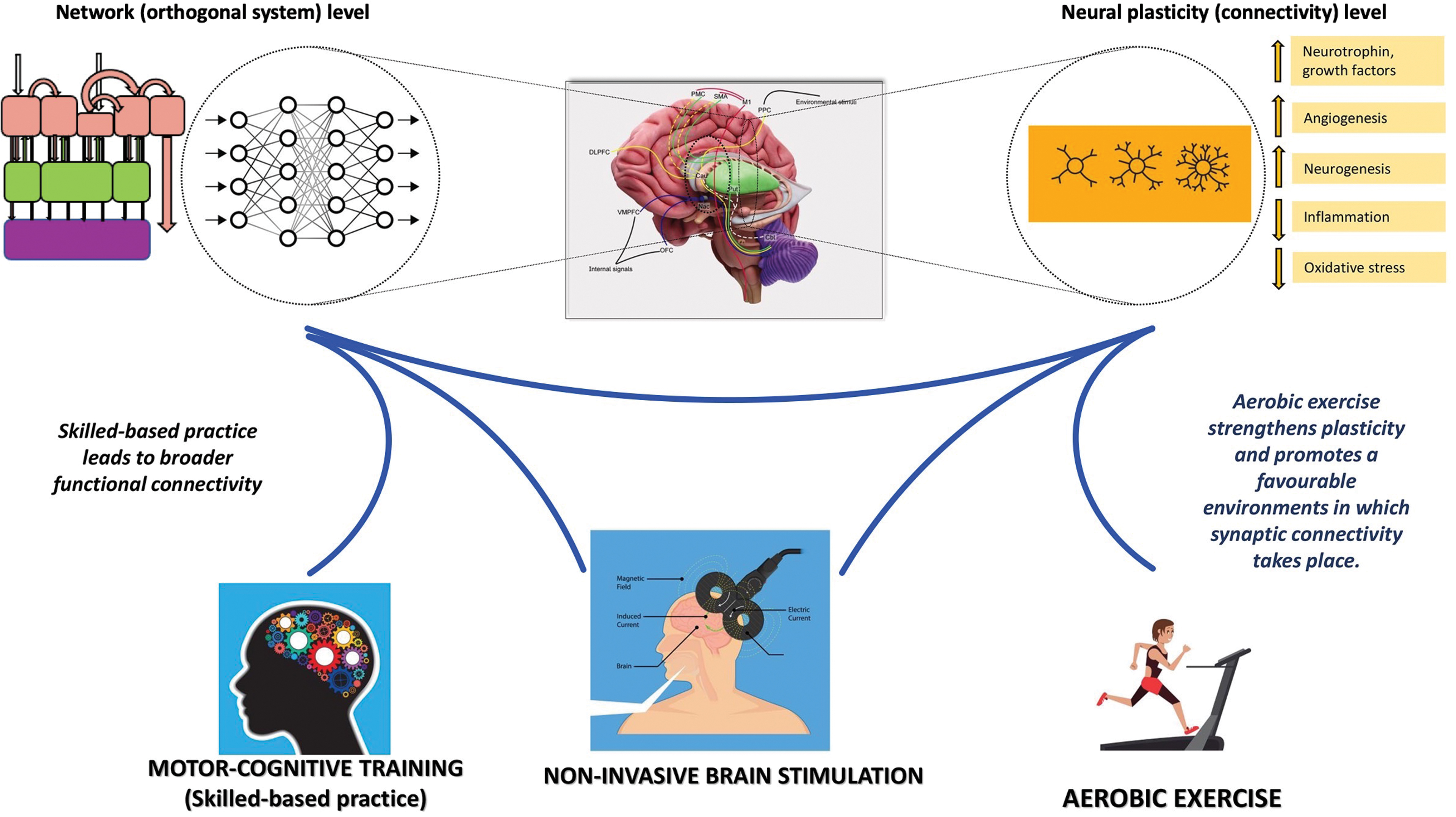
Rehabilitation for movement disorders. Skill-based practice merged with aerobic exercise and the use of NIBS can steer neural plasticity, modulate the activity of cortical–subcortical–cerebellar networks, and enhance retention of motor skills. See the text for details. NIBS, noninvasive brain stimulation techniques. Color images are available online.
Footnotes
Authors' Contributions
Research data for the article: D. Ferrazzoli, P. Ortelli, and L. Sebastianelli. Substantial contribution to discussion of content: D. Ferrazzoli, P. Ortelli, D. Volpe, A. Cucca, V. Versace, R. Nardone, and L. Saltuari. Writing: D. Ferrazzoli and P. Ortelli. Review/editing before submission: D. Ferrazzoli, P. Ortelli, and D. Volpe.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No companies, grants, or applications have financed this article.