
Abstract
Background:
The endogenous cannabinoid system modulates inflammatory signaling in a variety of pathological states, including traumatic brain injury (TBI). The selective expression of diacylglycerol lipase-β (DAGL-β), the 2-arachidonylglycerol biosynthetic enzyme, on resident immune cells of the brain (microglia) and the role of this pathway in neuroinflammation, suggest that this enzyme may contribute to TBI-induced neuroinflammation. Accordingly, we tested whether DAGL-β−/− mice would show a protective phenotype from the deleterious consequences of TBI on cognitive and neurological motor functions.
Materials and Methods:
DAGL-β−/− and -β+/+ mice were subjected to the lateral fluid percussion model of TBI and assessed for learning and memory in the Morris water maze (MWM) Fixed Platform (reference memory) and Reversal (cognitive flexibility) tasks, as well as in a cued MWM task to infer potential sensorimotor/motivational deficits. In addition, subjects were assessed for motor behavior (Rotarod and the Neurological Severity Score assays) and in the light/dark box and the elevated plus maze to infer whether these manipulations affected anxiety-like behavior. Finally, we also examined whether brain injury disrupts the ceramide/sphingolipid lipid signaling system and if DAGL-β deletion offers protection.
Results:
TBI disrupted all measures of neurological motor function and reduced body weight, but did not affect body temperature or performance in common assays used to infer anxiety. TBI also impaired performance in MWM Fixed Platform and Reversal tasks, but did not affect cued MWM performance. Although no differences were found between DAGL-β−/− and -β+/+ mice in any of these measures, male DAGL-β−/− mice displayed an unexpected survival-protective phenotype, which persisted at increased injury severities. In contrast, TBI did not elicit mortality in female mice regardless of genotype. TBI also produced significant changes in sphingolipid profiles (a family of lipids, members of which have been linked to both apoptotic and antiapoptotic pathways), in which DAGL-β deletion modestly altered levels of select species.
Conclusions:
These findings indicate that although DAGL-β does not play a necessary role in TBI-induced cognitive and neurological function, it appears to contribute to the increased vulnerability of male mice to TBI-induced mortality, whereas female mice show high survival rates irrespective of DAGL-β expression.
Introduction
The understanding of traumatic brain injury (TBI) being a simple acute event has evolved in conceptualization to reflect a chronic disease state originating from the initial traumatic insult. The learning and memory deficits commonly seen in clinical TBI cases frequently persist for decades after injury. 1 In addition to the initial mechanical disturbance of cellular membranes and axons, TBI perpetuates adaptive inflammatory immune responses that prompt a continued state of chronic inflammation. 2 The circulating proinflammatory mediators dysregulate the cellular underpinning of learning and memory, such as changes to the expression and composition of AMPA receptors, 3 promotion of neuronal hyperexcitability, 4 and impairing long-term potentiation induction and maintenance. 5 Inflammation can also be observed in humans for years after injury 6 as well as chronically in murine TBI models. 7 As such, chronic neuroinflammation is widely linked to further neurodegeneration and specifically learning and memory deficits of TBI.
Our understanding of the endocannabinoid (eCB) system has also evolved, from that of a prohomeostatic neurotransmitter system to include its contributions toward inflammatory eicosanoid production. 8 In particular, the primary biosynthetic enzymes of the most abundant eCB in brain, 2-arachidonylglycerol (2-AG), diacylglycerol lipase (DAGL)-α and -β, play distinct roles in the central nervous system (CNS) and periphery. DAGL-α is highly expressed on neurons and astrocytes in the CNS, and DAGL-β is expressed on microglia 9 and macrophages. The observations that DAGL-β disruption attenuates proinflammatory responses reveal its role in regulating inflammation.10,11 As such, manipulating proinflammatory pathways by blocking DAGL-β may have beneficial effects on TBI learning and memory outcomes.
The distinct cellular expression of the 2-AG biosynthetic enzymes and their unique roles in physiological function, renders manipulation of DAGL-β a particularly attractive therapeutic neuroinflammatory target for TBI. Unlike DAGL-α disruption, DAGL-β deletion does not impair eCB-mediated short-term synaptic plasticity.12,13 DAGL-α deletion leads to a 90% reduction of brain 2-AG,12,14 with concomitant profound adverse phenotypic consequences, such as memory impairment, decrements in neurogenesis, and changes in neuronal excitability in mice,15,16 as well as impairing AA signaling and function, including membrane fluidity and regulation of ion channels. 17 Conversely DAGL-β−/− mice show either a 50% reduction 12 or no change10,18,19 in whole-brain 2-AG, suggesting this enzyme plays a smaller, cell-specific role in establishing bulk 2-AG levels than DAGL-α (consistent with the observation that microglia composes only 5–12% of CNS cells 20 ).
Sphingolipids are another lipid family that plays important roles in cellular survival. Sphingolipids are ubiquitous components of all membranes that generate bioactive metabolites, including ceramide, which have been linked to apoptotic pathways and TBI-induced cellular dysfunction.21–24 Accordingly, TBI may activate acidic sphingomyelinase, which cleaves sphingomyelin to ceramide (also the precursor for monohexosylceramide by glycosylation). Formation of sphingosine (So) from degradation of ceramide further participates in mitochondria dysfunction and brain function impairment in TBI. 22
Given the expression of DAGL-β in the brain,12,25 its role as a key metabolic hub regulating proinflammatory lipid networks, 10 and given its disruption perturbs microglia eCB-eicosanoid crosstalk, the present study investigated the role of DAGL-β on TBI-induced disruption of learning and memory and motor function. We also examined whether TBI would elevate sphingolipids and if DAGL-β deletion offers protection. Because selective and brain-permeable DAGL-β inhibitors are yet to be optimized, 26 we utilized DAGL-β−/− and -β+/+ mice to examine whether nullifying this enzyme would offer protection from TBI-induced functional deficits.
Materials and Methods
Animals
Adult male DAGL-β−/−, and -β+/+ mice on a mixed 99% C57BL/6 (50% J [Jackson Laboratories, Bar Harbor, Maine], 50% N [Charles River, Wilmington, MA, USA]), and 1% 129/SvEv background, as previously described, 10 (females were included in the mortality experiment only). All mice (age 8–10 weeks) were pair-housed under a 12-h light/12-h dark cycle (0600–1800 h), at a constant temperature (22°C) and humidity (50–60%), with food and water available ad libitum. All experiments were conducted in accordance with the National Institute of Health (NIH) Guide for the Care and Use of Laboratory Animals (NIH Publications No. 8023, revised 1978), and were approved by the Virginia Commonwealth University Institutional Animal Care and Use Committees.
Craniectomy and induction of TBI
A craniotomy and a left lateral fluid percussion TBI were performed 27 (behavioral assessment experiment: 1.94±0.1 atmospheres (atm), mortality experiments: 2.0±0.1 and 2.17±0.1 atm). Immediately following the delivery of the injury, mice were placed on their backs to assess the righting reflex, which was scored as the duration of time to right themselves.
Mortality
Instances of mortality occurred within 2 min following the initial injury, otherwise mice survived until the end of the 34-day experiment. Therefore, experiments investigating mortality at increased injury severity, applied a 2 min postinjury cutoff.
Morris water maze assessments
A Morris water maze (MWM) (circular, galvanized steel tank, 1.8 m diameter, 0.6 m height) Fixed Platform task was used to assess reference memory and a Reversal task assessed cognitive flexibility. 27 A cued task assessed mice for potential sensorimotor/motivational deficits (see Fig. 1A for experimental timeline).
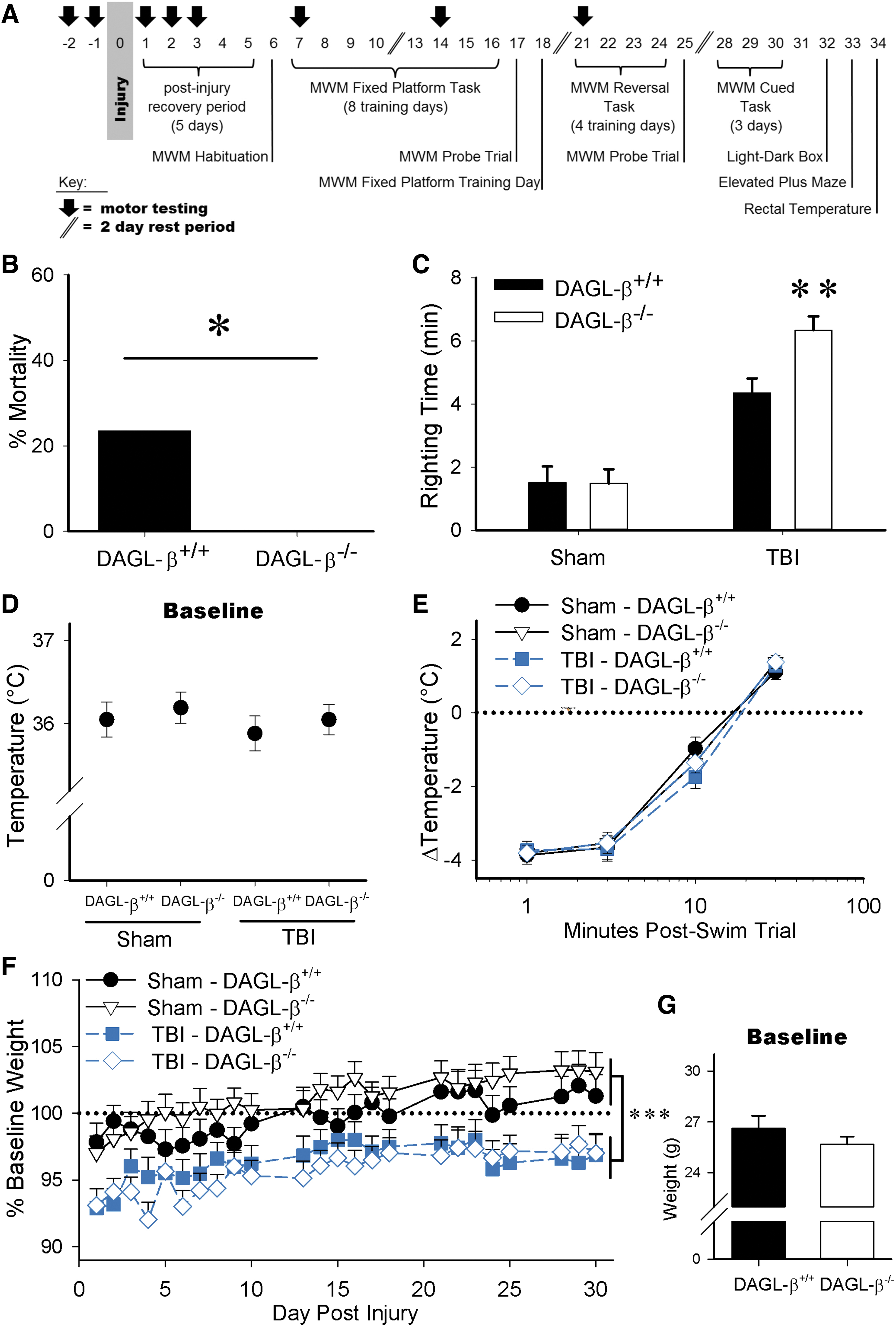
DAGL-β deletion improves TBI acute survival but increases injury-induced righting times, with no impact on body temperature or TBI-induced weight loss (Sham-DAGL-β+/+ n=12, Sham-DAGL-β−/− n=15, TBI-DAGL-β+/+ n=12, TBI-DAGL-β−/− n=16).
Neurological motor assessments
Mice were trained in assays of neurological motor impairment, Rotarod (IITC Life Science Rota-Rod, Woodland Hills, CA, USA) and Neurological Severity Score (NSS), 27 2 days before injury, and 1, 2, 3, 7, 14, and 21 days postinjury. In the NSS, one point is awarded for failing to perform a particular task, and no points for succeeding. The points of each of the 10 tasks are summed, in which a score of 0 represents a naive uninjured mouse, whereas a maximal score of 10 indicates severe neurological dysfunction.
Tests of affective behavior
Mice were assessed for affective behavior using the light/dark box and the elevated plus maze (EPM) on day 32 and 33 postinjury, respectively. The light/dark box consisted of an enclosed dark box (36×10×34 cm) with an opening (6×6 cm) to a larger brightly lit area (36×21×34 cm). After a 1-h acclimatization to the testing room, mice were placed in the light side of the apparatus to explore freely for 5 min, tracked by ANY-maze (San Diego Instruments, Inc., San Diego, CA, USA). The EPM (Hamilton-Kinder, Poway, CA, USA) consisted of a 60-cm-high platform. A central area (5×5 cm) allowed access to each of four arms, two (35×5 cm) enclosed by 15-cm-high walls, two arms had no walls. After 1 h acclimatization to the testing room, mice were placed in the central area facing an open arm to explore freely for 5 min. Photocell arrays connected to a MotorMonitor® system (Hamilton-Kinder) recorded movement.
Lipidomic analysis
Lipids were extracted from male mice ipsilateral half brain and lung tissue (collected at 2 min post TBI and flash frozen in liquid nitrogen), and quantified by means of liquid chromatography–electrospray ionization–tandem mass spectrometry (4000 QTRAP; AB Sciex, Concord, Ontario, Canada) as previously described. 28
Experimental design and statistical analysis
Behavioral data are presented as mean±standard error of the mean. The criterion for significance in all experiments was set at p<0.05, and all analyses were conducted using IBM SPSS Statistics 22 for Windows (IBM Software, New York, NY, USA). All changes in body temperature, % baseline weight, MWM Fixed Platform and Reversal acquisition, MWM swim speed, Rotarod, and NSS data were analyzed using a mixed factor analysis of variance (ANOVA) (°C, grams, distance, cm/s, and latency measures). All probe trial and cued task analyses, righting time, baseline temperature, light/dark box, EPM, and lipidomics data were analyzed using a two-way ANOVA (% time in quadrant, % time in outer ring, cm/sec, temperature, time, entries, and distance measures). Baseline weight was analyzed by an independent group's t-test. All mortality analyses were conducted using a Chi-square analyses by each injury severity.
Results
DAGL-β deletion improved TBI acute survival and increased injury-induced righting times, with no impact on body temperature or TBI-induced weight loss
Male DAGL-β−/− mice showed an unexpected significant reduction in TBI-induced acute mortality (i.e., immediately following injury) compared with DAGL-β+/+ mice [X2(1, N = 28)=4.53, p=0.033; Fig. 1B]. However, these mice took longer to recover from TBI-induced loss of righting than male DAGL-β+/+ mice (p=0.003), revealed by a significant interaction between injury and time [F(1,51)=4.63, p=0.036; Fig. 1C].
Both genotypes had nearly identical body temperatures (Fig. 1D). Following the first 2-min swim in the MWM, body temperatures substantially dropped and gradually recovered [main effect of time; F(3,153)=795, p=0.000, Fig. 1E] irrespective of injury or genotype. Posthoc analyses revealed hypothermic responses at 1 min (p=0.000), 3 min (p=0.000), and 10 min (p=0.000) postswim compared with 30 min.
TBI produced a significant reduction in body weight [F(1,51)=16.4, p<0.0001; Fig. 1F], but weight increased by postinjury days 13 through 30 compared with postinjury day 1 (consecutively; p=0.009, p<0.0001, p=0.001, p<0.0001, p=0.001, p=0.002, p<0.0001, p<0.0001, p<0.0001, p=0.038, p=0.002). No pre-existing body weight differences were seen between DAGL-β+/+ and DAGL-β−/− mice at baseline (day 0 presurgery/injury) (p=0.2605, T-test; Fig. 1G).
TBI impedes MWM task performance regardless of DAGL-β deletion
In MWM Fixed Platform acquisition, TBI mice had longer swim distances to the platform than sham mice [F(1,51)=0.237, p<0.0001; Fig. 2A], irrespective of DAGL-β deletion (p=0.397). TBI mice also swam slower than sham mice [F(1,51)=8.69, p=0.005, Fig. 2B], although all mice had significantly reduced swim distances [F(7,357)=32.2, p<0.0001] on Fixed Platform days 2 through 8 (all p<0.0001). The probe trial test revealed that TBI mice spent a reduced percentage of time in the target quadrant [F(1,51)=8.33, p=0.006; Fig. 2C], and had reduced platform location entries [F(1,51)=6.25, p=0.016; Fig. 2E] compared with sham mice, whereas DAGL-β deletion did not affect any of these indices. TBI did not however affect distance to the prior platform location (p=0.126; Fig. 2D). TBI also altered probe trial search strategy in which TBI mice spent significantly more time in the MWM outer ring than sham mice [F(1,51)=5.87, p=0.019, ANOVA; Fig. 2F], as well as used circular nonspatial search strategies (Fig. 2G). The absence of significant differences in cued task performance (p=0.572; Fig. 2H, p=0.692; Fig. 2I) suggests that TBI did not produce overt sensorimotor or motivational alterations. Similarly, TBI disrupted cognitive flexibility as assessed in the reversal task irrespective of DAGL-β deletion (Supplementary Fig. S1 and Supplementary Data).
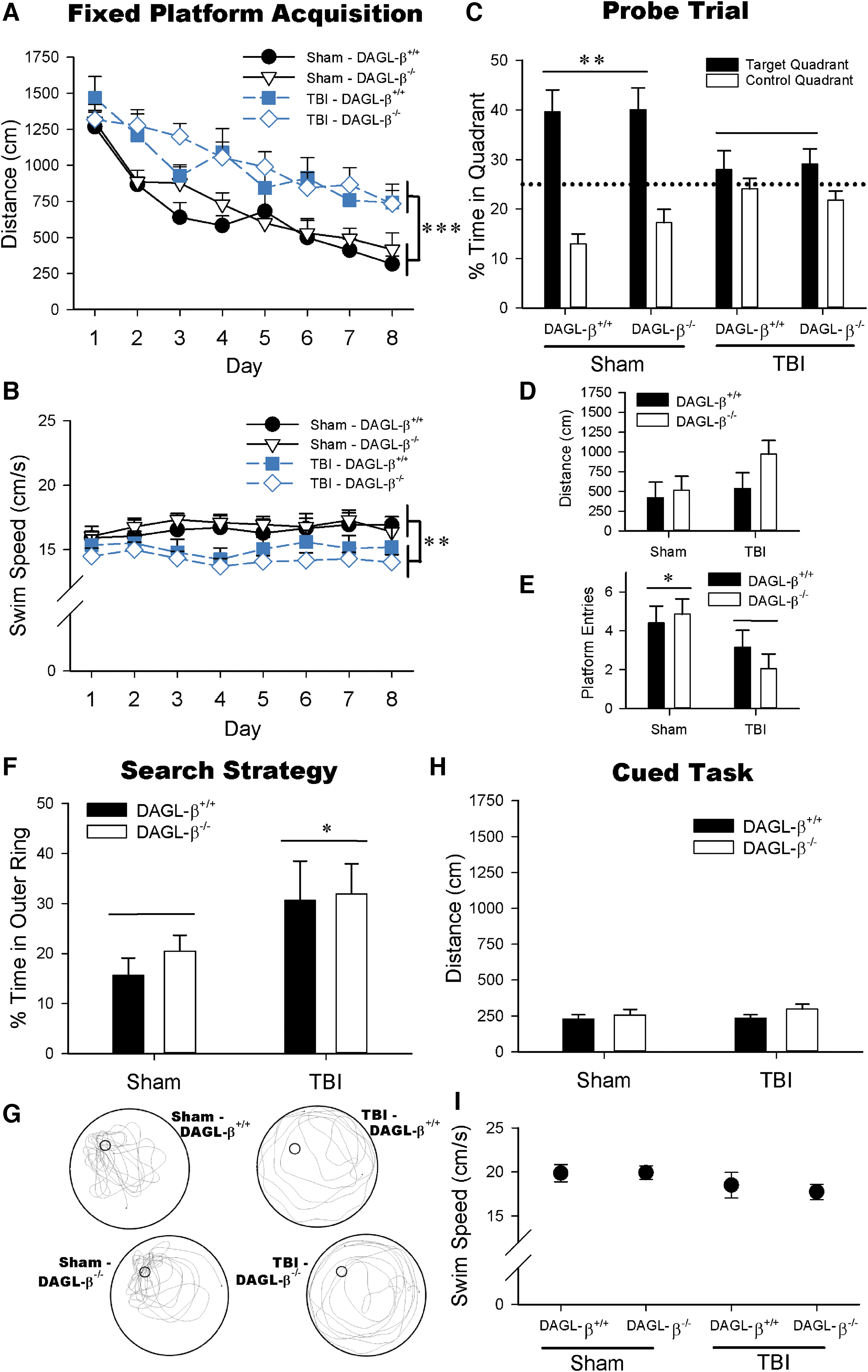
TBI produces reference memory deficits, irrespective of DAGL-β deletion (Sham-DAGL-β+/+ n=12, Sham-DAGL-β−/− n=15, TBI-DAGL-β+/+ n=12, TBI-DAGL-β−/− n=16).
TBI impairs motor performance irrespective of DAGL-β deletion.
TBI reduced latencies to fall from the Rotarod [F(7,357)=6.20, p<0.0001; Fig. 3A] on postinjury days 1 (p<0.0001), 2 (p<0.0001), and 3 (p<0.0001) compared with sham mice, which resolved by day 7 (Fig. 3A). However, DAGL-β deletion did not protect against this neurological motor impairment (nonsignificant: three-way interaction [p=0.494], two-way interactions with genotype [day; p=0.647, injury; p=0.846]), or main effect of genotype [p=0.506]).

TBI produces neurological motor impairments, irrespective of DAGL-β deletion (Sham-DAGL-β+/+ n=12, Sham-DAGL-β−/− n=15, TBI-DAGL-β+/+ n=12, TBI-DAGL-β−/− n=16).
Similarly, TBI worsened NSS scores [F(6,306)=8.04, p<0.0001] on postinjury days 1 (p=0.002), 2 (p=0.028), and 7 (p=0.010) compared with sham mice, which resolved by day 14 postinjury (Fig. 3B). Again, these TBI-induced deficits occurred irrespective of genotype indicating that DAGL-β deletion did not offer protection from motor impairments (nonsignificant: three-way interaction [p=0.962], two-way interactions with genotype [day; p=0.627, injury; p=0.624], or main effect of genoptype [p=0.279]).
Neither TBI nor DAGL-β deletion affects behavior in the light/dark box or EPM
As shown in Supplementary Figure S2 and Supplementary Data, neither TBI nor genotype impacted light/dark box or EPM performance. However, diazepam produced anxiolytic phenotypes in both genotypes during light/dark box and EPM performance.
Male DAGL-β−/− mice exhibit a survival-protective phenotype
Additional experiments examined whether the unexpected survival phenotype in male DAGL-β−/− mice would persist with an increased magnitude of injury and if female mice would show a similar phenotype. Male DAGL-β−/− mice showed decreased mortality rates compared with male DAGL-β+/+ mice with increased injury forces of 2.0 atm [X2(1, N=31)=4.31, p=0.038] and 2.17 atm [X2(1, N = 28)=4.09, p=0.043; Fig. 4A]. However, female mice showed high survival rates (2.0 atm; DAGL-β+/+ 93%, DAGL-β−/− 100%, 2.17 atm; DAGL-β+/+ 86%, DAGL-β−/− 100%) regardless of genotype at both 2.0 atm [X2(1, N=30)=1.03, p=0.309] or 2.17 atm [X2(1, N=28)=2.15, p=0.142; Fig. 4B], demonstrating a sex-dependent protective effect.
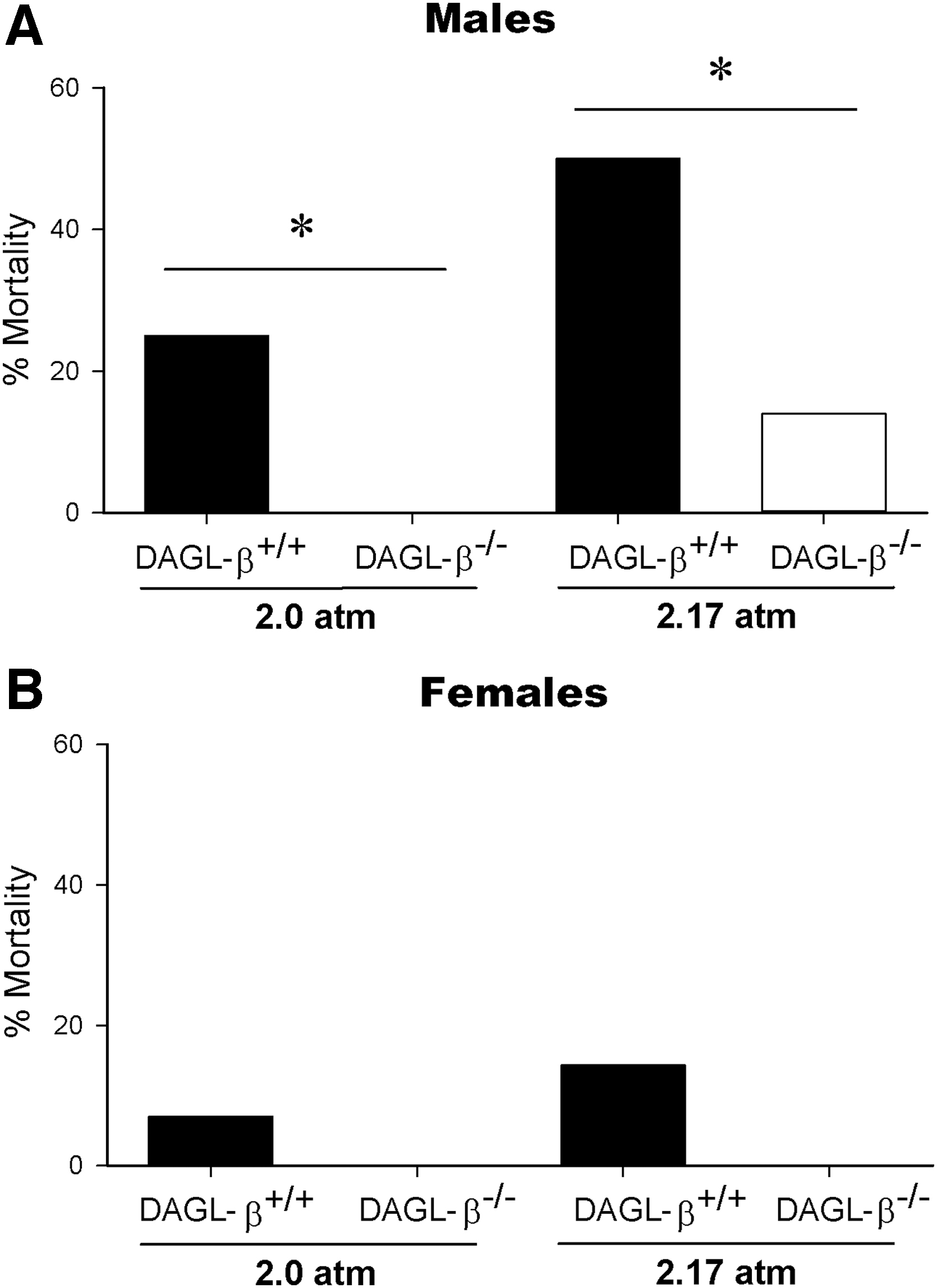
Male DAGL-β−/− mice exhibit a survival-protective phenotype, which persists at increased injury severity. The survival-protective phenotype of DAGL-β−/− mice persisted at increased injury severity in male mice (male TBI mice; [2.0 atm]-DAGL-β+/+ n=16, [2.0 atm]-DAGL-β−/− n=15, [2.17 atm]-DAGL-β+/+ n=14, [2.17 atm]-DAGL-β−/− n=14), whereas being female was generally protective (female TBI mice; [2.0 atm]-DAGL-β+/+ n=15, [2.0 atm]-DAGL-β−/− n=15, [2.17 atm]-DAGL-β+/+ n=14, [2.17 atm]-DAGL-β−/− n=14).
Evaluation of TBI and DAGL-β deletion on levels of ceramide and sphingolipid species in brain and lung
We examined the consequences of acute TBI in DAGL-β−/− and DAGL-β+/+ mice on levels of ceramide, sphingomyelin, and monohexosylceramide species. Acute TBI increased most ceramide species C16:0 [F(1,28)=139.8, p<0.0001], C24:1 [F(1,28)=106.7, p<0.0001], C24:0 [F(1,28)=33.7, p<0.0001], and C26:1 [F(1,28)=118.2, p<0.0001], whereas significantly decreasing ceramide species C20:0 0 [F(1,28)=6.49, p=0.017], and C22:0 [F(1,28)=17.4, p<0.0001], in both genotypes (Fig. 5A). Interestingly, TBI lowered C18:0 ceramide in DAGL-β−/− mice compared with TBI DAGL-β+/+ mice [F(1,28)=5.22, p=0.030; Fig. 5A]. Of note, this sphingolipid has been implicated in lethal mitophagy, 29 which is relevant given that decreased energy production from TBI-induced mitochondrial dysfunction aggravates cell damage and cell death when a high energy demand exists for cellular repair.

Acute TBI produces robust changes in brain sphingolipid profiles with impact on specific species by DAGL-β deletion (all groups n=8). Sphingolipids were extracted from ipsilateral brain of DAGL-β+/+ and DAGL-β−/− mice 2 min post-TBI.
DAGL-β−/− mice showed significant reductions in major brain monohexosylceramide species [i.e., C18:0, C20:0, C22:0, C24:1, and C24:0, F(1,28)=12.9, p=0.001; F(1,28)=11.5, p=0.002; F(1,28)=13.1, p=0.001; F(1,28)=4.54, p=0.042; F(1,28)=24.7, p<0.0001, respectively] compared with DAGL-β+/+ mice after injury (p<0.0001, p<0.0001, p<0.0001, p=0.005, and p<0.0001, respectively; Fig. 5B). TBI also significantly decreased monohexosylceramide at chain lengths; C16:0 [F(1,28)=169.3, p<0.0001] and C26:1 [F(1,28)=100.9, p<0.0001], but increased C18:1 monohexosylceramide [F(1,28)=105.8, p<0.0001].
TBI significantly decreased all brain sphingomyelin species (Fig. 5C); C16:0 [F(1,28)=13.4, p=0.001], C18:1 [F(1,28)=45.8, p<0.0001], C18:0 [F(1,28)=17.03, p<0.0001], C20:0 [F(1,28)=162.8, p<0.0001], C22:0 [F(1,28)=294.3, p<0.0001], C24:1 [F(1,28)=12.1, p=0.002], C24:0 [F(1,28)=167.8, p<0.0001], and C26:1 [F(1,28)=275.3, p<0.0001], irrespective of genotype. Injury also significantly increased levels of So [F(1,28)=15.3, p=0.001; Fig. 5D] and sphinganine (dihydrosphingosine) [F(1,28)=74.6, p<0.0001; Fig. 5E], but did not significantly affect levels of the prosurvival sphingolipid metabolite sphingosine-1-phospate (p=0.740; Fig. 5F) in the brain.
As shown in the lung (Fig. 6A), acute TBI produced a modest decrease in one ceramide species, C26:1 [F(1,36)=5.89, p=0.020], whereas DAGL-β−/− mice had lower levels of C20:0 ceramide than DAGL-β+/+ mice [F(1,36)=4.85, p=0.034]. No change was evident in lung monohexosylceramide in response to acute TBI (Fig. 6B); however, TBI significantly decreased lung sphingomyelin species C16:0 [F(1,36)=10.5, p=0.003], C20:0 [F(1,36)=5.68, p=0.023], C22:0 [F(1,36)=8.66, p=0.006], and C26:1 [F(1,36)=7.91, p=0.008; Fig. 6C] similar to the brain.

Acute TBI produces modest changes in lung sphingolipid profiles with no impact on specific species by DAGL-β deletion (all groups n=8). Sphingolipids were extracted from lung of DAGL-β+/+ and DAGL-β−/− mice 2 min post-TBI.
Discussion
The present study tested whether DAGL-β deletion protects against TBI-induced learning and memory deficits. In this study, we report that TBI disrupted mouse Fixed Platform and Reversal task performance accompanied by nonspatial search strategies, irrespective of DAGL-β deletion. Thus, deletion of this enzyme did not provide protection from TBI-induced spatial learning and memory impairments. Additionally, DAGL-β deletion did not provide protection from TBI-induced neurological motor function deficits and reductions in body weight. Neither TBI nor genotype affected body temperature regulation or performance in a common assay used to infer anxiety. However, DAGL-β deletion produced a surprising survival-protective phenotype in male mice, which persisted at increased injury severities. In contrast, female mice did not display injury-related mortality regardless of genotype. As mortality is not a frequently explored outcome measure in preclinical studies of TBI, these findings are novel. TBI also produced significant acute changes in sphingolipid profiles and DAGL-β deletion reduced levels of specific sphingolipid species that might have unique functions in pathological processes leading to injury-induced mortality in male mice.
The unexpected finding that DAGL-β disruption produced a survival-protective phenotype in male mice, suggests a novel role of this enzyme in acute TBI pathology. TBI-induced mortality reflects an infrequently explored preclinical outcome measure. The lack of effects of DAGL-β deletion on fixed platform and reversal MWM performance suggests that the survival-protective phenotype did not increase sensitivity or resistance to functional impairment (despite their increased righting times compared with to -β+/+ mice). Whereas one study showed decreased TBI-related mortality in individuals with a positive Δ 9 -tetrahydrocannabinol urine screen, 30 another study reported that preinjury cannabis use did not improve survival in trauma patients, but it improved outcomes in patients with severe TBI. As the observed mortality in the present study occurred within a 2-min postinjury window, secondary injury mechanisms relevant to such an acute time scale should be considered. For example, glutamate excitotoxicity occurs within minutes following injury,31,32 as does the generation of ceramide through the hydrolysis of sphingomyelin catalyzed by sphingomyelinases. 33 Ceramide is linked not only to glutamate toxicity but also to mitochondrial dysfunction, and promotes apoptotic and autophagic cell death.34,35 This raises the possibility that TBI leads to activation of acidic sphingomyelinase, which increases levels of ceramide and its metabolite, which then contributes to mitochondrial dysfunction. 22 Consistent with previous studies,21–23 we observed a robust TBI-induced degradation of sphingomyelin and elevation of ceramide and so. Our data also support an emerging idea that different ceramide species have unique functions.34,35 DAGL-β−/− mice showed a significant reduction in TBI-induced C18:0 ceramide. Notably, this ceramide species plays a critical role in lethal mitophagy, 29 and activation of protein phosphatase 2 (PP2A) and impairing survival pathways, 36 which implicates its possible contribution to the survival phenotype of male DAGL-β−/− mice reported in this study. Moreover, TBI increased and DAGL-β deletion decreased monohexosylceramides (glucosylceramide and galactosylceramide) levels. Although the functions of monohexosylceramide in TBI remain to be examined, it is well established that the respective brain accumulation of glucosylceramide and galactosylceramide in Gauche and Krabbe disease patients leads to massive neuronal cell death.37,38 Thus, it is tempting to speculate that increases of monohexosylceramide play an important yet unappreciated role in the pathology of TBI.
The finding that female DAGL-β+/+ mice showed high TBI survival rates is consistent with female rodents being generally protected from TBI-induced mortality compared with males.39,40 The high rate of survival in female mice indicates that DAGL-β plays a sex-dependent role. Although the mechanisms underlying female protection from TBI mortality remain unclear, in this study, we consider several possibilities. Preclinical studies report that female mice demonstrate a biphasic proinflammatory response, whereas male mice show a prolonged single-phase increase in inflammatory mediators, 41 although early acute 2-min time points are rarely evaluated. Inflammation resolution profiles also differ, with female mice showing a delayed peak of anti-inflammatory mediators, whereas male mice again show a single phase. 41 Preclinical studies also show a potential hormonal role driving sex differences in TBI mortality profiles. Umeano et al. showed similar mortality rates of ovariectomized female mice as males suggesting that female gonadal hormones may influence murine TBI mortality outcomes. While administration of estrogen and progesterone to male mice shows neuroprotection in functional and molecular endpoints,42–45 no known rodent work to date has linked mortality protection by these hormones. Thus, studies examining TBI survival in ovariectomized female DAGL-β−/− mice and wild-type male mice, with estrogen/progesterone versus vehicle pretreatment, might provide insight into understanding sex differences in mouse TBI mortality found in the present study. Furthermore, the additional analysis of female sphingolipid profiles following TBI would likely also be informative. Female mortality in clinical populations differs to male only when cohorts are evaluated by age, with pubescent 46 and postmenopausal women47,48 showing mortality protection, both being life cycle times of estrogen unbalanced by progesterone. Furthermore, two large randomized clinical trials found no protection of progesterone.49,50 As such, the mechanisms by which females are protected from TBI mortality compared with males strongly suggest the involvement of estrogen.
While the present results support acute inflammatory effects (2 min) on TBI mortality and sphingolipid modulation, global chronic behavioral deficits and associated pathology were unaltered. Even though the disease state induced after a TBI is composed of heterogeneous secondary injury mechanisms, chronic neuroinflammation has been a singularly popular neuroprotective research target, perhaps given the rich number of neuroinflammatory sites as well as the inflammatory temporal profile providing intervention opportunities following the primary insult. Yet thus far, all late-phase clinical trials have failed to yield an effective anti-inflammatory neuroprotective treatment.51,52 The modulation of neuroinflammation in murine models of TBI have produced mixed success, with high-dose anti-inflammatories (non-steroidal anti-inflammatories and glucocorticosteroids) exacerbating learning and memory deficits,53–55 and isolated stand-alone drugs targeting neuroinflammation (antibiotics, COX-1 inhibitors, a plant-based alkaloid, and an apolipoprotein mimetic peptide) attenuating memory impairment.56–59 The mixed results of anti-inflammatory drug targets in preclinical research and the failed translation to clinical learning and memory protection is perhaps a consequence of the limitations of animal models as well as the necessity to promote the beneficial effects of acute neuroinflammation, while minimizing detrimental chronic effects. Intervention timing post-TBI is an important consideration in the present results given that a global DAGL-β knockout mouse does not allow for temporal intervention manipulation. Nonetheless, DAGL-β expression is upregulated during an activated microglial phenotype, 60 suggesting that this enzyme may indeed play a temporal dependent role with regard to inflammation pathology, which fits the presently observed acute postinjury effects. The current constitutive genetic deletion of DAGL-β may, therefore, mask time-dependent protective effects of DAGL-β disruption against TBI-induced memory deficit. Future experiments using pharmacological tools would be valuable to manipulate the timing of DAGL-β disruption, but are currently challenging given the lack of availability of selective and brain-penetrant DAGL-β inhibitors. Furthermore, 2-AG is a potent cannabinoid type 2 (CB2) receptor agonist, 61 consequently DAGL-β gene deletion may also result in decreased activation of microglial or macrophage CB2 receptors. Given that CB2 receptors promote inflammation resolution, reduced CB2 receptor signaling could offset any presumed beneficial effects of reduced AA metabolites. Indeed, the inhibition of 2-AG catabolic enzymes, monoacylglycerol lipase, and alpha/beta-hydrolase domain 6, decreased neuroinflammation and improved cognitive function in TBI mouse models.62,63
Although DAGL-β deletion was not protective against TBI-induced spatial learning and memory deficits, further work to investigate how this enzyme affects neuroinflammation and TBI outcome remains pertinent as TBI patients are predisposed to develop other neurological pathologies, such as Alzheimer's disease, 64 thought to be a product of prolonged neuroinflammation. 65 Moreover, the present study raises the provocative possibility that DAGL-β activity influences acute mortality following TBI in male subjects, implicating an important sex-dependent role of this enzyme in the very early phases of TBI pathology.
Footnotes
Disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the VCU Lipidomics/Metabolomics Core, the National Institutes of Health (NIH-NCI) Cancer Center Support Grant P30 CA016059 to the Massey Cancer Center, NIH shared resource grant (S10RR031535), NIH [Grant NS093990], and NIH [Grant P30DA033934] (to A.H.L.); NIH [Grant DA033760] (to B.F.C.); NIH [Grant 1F31NS095628] (to L.D.OB.); NIH [Grant K99HD096117] (to J.N.); NIH [Grant NS57758] (to T.M.R.); NIH [Grant GM043880] (to S.S.); and NIH [Grant NS56247] (to L.L.P.). Start-up funds from the VCU School of Pharmacy also supported this work.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.