
Abstract
Rationale:
The slowing of disease progression in dementia in the early stages of diagnosis is paramount to improving the quality of life for those diagnosed and their support networks. Accumulating evidence suggests that CBD, a constituent of Cannabis sativa, is associated with neuroprotective, neuroendocrine, and psychotherapeutic effects, suggesting that it may be beneficial to dementia treatment. However, no published human study to date has examined this possibility. This trial aims to determine whether daily treatment with CBD over a 12-week period is associated with improved neurobiological, behavioral, and psychological outcomes in individuals living with early-stage dementia.
Methods:
Sixty participants with early-stage dementia will be recruited for a randomized, double-blind, placebo-controlled clinical trial. Participants will be randomized into either 99.9% pure CBD or placebo treatment conditions and administered two capsules per day for 12 weeks. Participants will commence a 200 mg/day dose for 2 weeks before escalating to 300 mg/day for the remaining 10 weeks. Neuroimaging and blood-based neuroendocrine profiles will be assessed at baseline and post-treatment. Psychological and behavioral symptoms will be assessed at baseline, 6 weeks, and post-treatment. Monitoring of health and side-effects will be conducted through weekly home visits.
Discussion:
This study is among the first to investigate the effects of isolated CBD in improving neuroanatomical and neuroendocrine changes, alongside psychological symptoms, during the early stages of dementia diagnosis. The outcomes of this trial have the capacity to inform a potential novel and accessible treatment approach for individuals living with early-stage dementia, and in turn, improve quality of life, prognoses, and treatment outcomes.
Trial Registration:
This trial has been registered with the Therapeutic Goods Administration (CT-2020-CTN-03849-1v2) and the Australian and New Zealand Clinical Trials Registry (ACTRN12621001364864).
Introduction
Dementia is a progressive syndrome characterized by cognitive, behavioral, and emotional changes. Key pathophysiological mechanisms in the early stages of dementia across some subtypes include neurodegeneration and associated changes in brain functioning and plasticity, 1 neuroendocrine dysregulation in various inflammatory and oxidative stress pathways,1,2 cognitive impairment, and behavioral and psychological changes. 3 People living with dementia and their support networks are often faced with complex and challenging treatment decisions at the time of diagnosis, resulting in increased distress and burden. Early interventions have the capacity to improve long-term prognoses, however, existing treatments are limited as they do not treat the pathophysiology of dementia or delay disease progression. 4 As such, people living with dementia are increasingly turning to alternative treatments, such as CBD. 5
CBD is a nonintoxicating phytochemical from Cannabis sativa that may be promising for therapeutic use in dementia due to its neuroprotective, neuroendocrine-regulating, and psychotherapeutic properties.6,7 With the recent down scheduling of CBD to an over-the-counter medication in Australia, 8 and the greater availability of prescribing and guidance information from regulatory agencies,8,9 there is a growing interest in and demand for CBD.10,11 However, there is a lack of clinical evidence indicating efficacy of CBD in older populations and in treating early-stage dementia, warranting investigations into its potential role as a therapeutic. 11
Much of the current evidence indicating support for CBD as a treatment for early-stage dementia is preclinical or demonstrated in other populations. In relation to neuroprotective effects, preclinical studies suggest that CBD protects against stress-induced cortical volume loss, 12 enhances neurogenesis in the hippocampus, 12 protects cerebral circulation and function during ischemic events, 13 and increases brain concentrations of adenosine and brain-derived neurotrophic factor (BDNF) to promote neuroprotection.12,13 Such effects have been linked to improved memory performance.14,15
Our research group demonstrated in cellular models of Alzheimer's that CBD is able to improve neural integrity, and promote dendritic structuring and neural growth. 16 In humans, we identified that CBD may also have neurorestorative effects, as long-term exposure to CBD in chronic cannabis users was associated with more normalized hippocampal volumes compared with those with long-term exposure to delta-THC 17 alone. In addition, our trial of 10–12 weeks CBD administration in chronic cannabis users resulted in increased hippocampal subfield volumes, associated with plasma CBD metabolite concentrations. 18
With respect to neuroendocrine effects, preclinical and human studies indicate that CBD can reduce neuroinflammation and oxidative stress by decreasing the production of proinflammatory cytokines, TNF-α and interleukin-67,19; proinflammatory adipokines, including leptin, 20 stress hormone cortisol, 21 and may be of relevance to proinflammatory and peripheral oxidative stress marker plasma serotonin,22,23 where reduced inflammation and oxidative stress may influence psychotherapeutic effects. 24
Indeed, preclinical studies administering CBD before laboratory stressors report reversed depressive, anxiety, and psychosis-related behaviors, such as low mood, learned helplessness, 7 fear responses, hyperlocomotion, 25 and poor cognition. 15 In humans, CBD administration is associated with improved mood, 26 reduced anxiety in generalized anxiety disorder, 27 and reduced psychotic symptomology in schizophrenia. 28 In chronic cannabis users, up to 12 weeks CBD administration was associated with reduced self-reported psychological symptom distress, depressive and anxiety symptom severity, and improved cognition. 29
More direct evidence is required to assess whether CBD has any real-world efficacy in early-stage dementia. 11 Most human trials investigating medicinal cannabinoids in dementia have to date examined products comprising THC only or combined CBD and THC.30–32 One human clinical trial investigating isolated oral CBD at 200–600 mg/day for 6 weeks to treat Alzheimer's Disease recently commenced in the United Kingdom, with primary outcomes, including acceptability and compliance of CBD, and secondary outcomes, including behavioral and psychological symptoms, cognition, and olfaction. 33
To our knowledge, no study to date has investigated isolated oral CBD in improving neuroanatomical and neuroendocrine changes alongside cognitive and psychological symptoms during the early stages of dementia diagnosis, particularly over a 12-week period. Considering emerging evidence, investigating the treatment efficacy of isolated CBD has the potential to inform a novel treatment that targets several facets of early-stage dementia, which have the capacity to improve quality of life.
Aims and Outcomes
The aim of this clinical trial is to determine whether daily treatment with CBD over a 12-week period is associated with improved neurobiological and psychological outcomes in individuals with early-stage dementia. Outcomes for this study include:
Primary outcomes (12 weeks after intervention commencement)
Change in brain structure and white matter connectivity, as measured by magnetic resonance imaging (MRI) scans and tractography, respectively.
Change in inflammatory and oxidative stress hormone concentrations (TNF-α, IL-6, leptin, cortisol, plasma serotonin), as measured in blood and enzyme-linked immunosorbent assay (ELISA).
Change in cognition, as measured by the Cognitive Functional Composite (CFC 34 ) and Frontal Assessment Battery (FAB 35 ).
Change in depression, as measured by the Geriatric Depression Scale (GDS 36 ) and anxiety, as measured by the Rating Anxiety in Dementia Scale (RAID 37 ).
Secondary outcomes (6 and 12 weeks after intervention commencement)
Change in quality of life, as measured by the Quality of Life-Alzheimer's Disease Scale (QOL-AD 38 ) and daily functional capacity, as measured by the Bristol Activities of Daily Living Scale (BADLS 39 ).
Change in neuropsychiatric symptoms and psychopathology, as measured by the Neuropsychiatric Inventory (NPI 40 ).
Change in appetite, eating behaviors, and eating patterns, as measured by the Appetite and Eating Behavior Questionnaire (APEHQ 41 ) and 3-Day Diet History Record (DHR).
Change in sleep patterns, as measured by the Sleep Disorders Inventory (SDI 42 ).
Change in agitation, as measured by the Brief Agitation Rating Scale (BARS 43 ), and aggression, as measured by the Rating Scale for Aggressive Behavior in the Elderly (RAGE 44 ).
Tertiary outcomes (6 and 12 weeks after intervention commencement)
Blood pressure, as measured in milligrams of mercury (mmHg) using an electronic sphygmomanometer.
Heart rate, as measured in average beats per minute (bpm) using an electronic sphygmomanometer.
Body mass index (BMI), as measured using a stadiometer (height in cm) and digital scale (weight in kg).
Waist circumference, as measured in centimeters (cm) at the halfway point between the lowest rib and the top of the hipbone during outbreath.
Methods
Trial design and setting
The study will utilize a double-blind, randomized, placebo-controlled design, where the study protocol (version 7) adheres to Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) guidelines (see Supplementary Data). 45 The study will be conducted at the Illawarra Health and Medical Research Institute (IHMRI) at the University of Wollongong (UOW) and Wollongong Diagnostics, Corrimal. Participation will involve three face-to-face time points at baseline, mid-way (6 weeks), and post-treatment (12 weeks). To reduce burden, baseline and post-treatment assessments will be conducted across 2 days. Weekly monitoring will assess symptoms and wellbeing through home visits from a registered nurse. Any modifications to the study protocol will be communicated with the trial research team, the Joint University of Wollongong and Illawarra Shoalhaven Local Health District Health and Medical Ethics Committee, and the Australian and New Zealand Clinical Trials Registry (ANZCTR).
Intervention
Medical grade 99.9% pure CBD will be formulated into gelatin oral capsules, containing either 100 or 200 mg CBD powder dissolved in SOFTISAN (Trigal Pharma Ltd.; subsidiary of BioSynthesis Pharma Group Ltd.). Placebo capsules will contain only SOFTISAN (Trigal Pharma Ltd.) and will appear identical to the CBD capsules in terms of appearance, flavor, and scent. Participants will administer one 100 mg capsule twice daily, morning and evening (total 200 mg/day) for the first 2 weeks. Capsule administration will commence the evening after baseline assessments are completed. The evening dose will be escalated to 200 mg in the third week of the trial, for a total 300 mg daily dose for the remaining 10 weeks, provided no significant adverse effects have been reported in the first 2 weeks.
Inclusion and exclusion criteria
Participants will be eligible for the study if they:
Are between 55 and 80 years of age; Are community dwelling, with a supporting person available to support them during the study; Have a clinician-confirmed primary diagnosis of early-stage dementia within 24 months of study participation; Have a clinician-endorsed Stage 3–5 rating on the Global Deterioration Scale
46
; and Can provide written informed consent. In line with previous literature, capacity for consent will be determined by a Mini Mental State Exam (MMSE
47
) score above 20.
48
Participants will be excluded on the basis of:
MRI contraindications, such as severe head injury, pace devices, coronary or periphery artery stents, cochlear implants, renal insufficiency, or claustrophobia;
Allergies to the excipient used in the formulation of the capsules (SOFTISAN);
Current impaired liver, kidney, or heart function;
Active suicidal ideation;
Intake of CBD, THC, or cannabis within 1 month before participation;
Prior extensive use of CBD, defined as use greater than weekly for more than 3 months in the 2 years before participation;
A history of recreational cannabis use, or other drugs of abuse, defined as more than three times per week for any continuous period of 6 months in duration in the year before participation;
Prior history of treatment for alcohol use disorder in the year before participation, or a current score of 8 or higher on the Alcohol Use Disorders Identification Test (AUDIT);
Any current or recent diagnoses of neurological disorders other than dementia, including epilepsy or Parkinson's disease;
Current use of medications that have clinically relevant interactions with the cytochrome P450 (CYP) 2C19 or CYP3A classes of liver enzymes, including antidepressants, antipsychotics, or anxiolytics;
Participation within any other clinical drug trials within 30 days of the study;
Anticipated surgical needs within 3 months of participation.
Recruitment
Sixty participants will be recruited through treating clinician referrals (general practitioners or dementia specialists from local private practices or hospitals), supporting persons, outpatient clinics, community groups, general media, and social media. Since a diagnosis of dementia requires confirmation by a specialist, if participants are sourced from anywhere other than a dementia expert, their diagnosis will be confirmed by the trial dementia experts. Interested parties will be provided with the participant information sheet outlining the study and its requirements, and will undergo an initial prescreening interview. Those who satisfy the eligibility criteria will be required to have their eligibility endorsed by their treating clinician. Retention of participants in the trial will be facilitated through weekly contact with participants and their supporting persons for screening, the three testing sessions, and weekly home visits for monitoring.
Consent
Participants will be required to provide written informed consent. Capacity for consent will be determined by a MMSE score above 20. 48 During the screening process, participants must be able to verbally explain the study and participation requirements to a satisfactory level to the research team and their treating clinician before enrollment in the study. Written informed consent will be obtained from participants and their supporting person during the baseline testing session, reconfirmed during the midpoint and post-treatment sessions, and checked at each stage of the data collection process (e.g., before biometric data collection, blood sampling).
Due to the potential for cognitive fluctuations over the trial period, the MMSE will be readministered at the midpoint of the trial (week 6) for potential changes in cognitive function and capacity to consent. Participants will be asked to explain the study and participation requirements again during weeks 3, 6, and 9 to indicate their ongoing understanding of study participation. If a participant's level of understanding changes, their participation in the trial will be reviewed in consultation with their treating clinician and the members of the research team.
Randomization and allocation concealment
In this double-blind trial, two senior investigators not directly involved in data collection will be unblinded to treatment allocations, whereas the participants, their supporting persons, and the remainder of the research team will be blinded to treatment condition allocations until study completion. Participants will be randomized into either pure CBD (N=30) or placebo (N=30) treatment conditions using a permuted block allocation strategy. A random number list using a 1:1 ratio allocation will be generated in Microsoft Excel by one of the unblinded researchers, which will sequentially match participant IDs (while balancing sex, age, and dementia severity as determined by MMSE scores across treatment arms) with unique batch codes to conceal the sequence of treatment allocation. Individual blinding allocations will be kept in a password-protected online server, with the password held only by an external researcher. Researchers involved in data collection will be informed of the batch code by the unblinded researchers for medication administration.
Adverse events, safety, and justification of dosage
CBD is nonintoxicating, is associated with a good safety profile and is well tolerated by humans at high doses with minimal adverse effects, including a lack of withdrawal symptoms. 49 However, in some individuals, CBD administration of longer than 6 weeks may cause fatigue, drowsiness, headaches, and mild gastrointestinal distress, including nausea, vomiting, or diarrhea.50–52 CBD has not previously been associated with adverse changes in physiological parameters such as blood pressure, nor psychological functioning. 53 Drug interactions have been reported between CBD and medications metabolized through the CYP 2C19 or CYP3A pathways in the liver, including antidepressants such as citalopram, escitalopram, and sertraline; antipsychotics such as clozapine; or anxiolytics such as diazepam and lorazepam; resulting in an increased risk of adverse events, including tiredness, gastrointestinal distress, and mild changes in hepatic function. 54
To maximize participant safety, a dose-escalation approach for capsule administration will be implemented. Dose-escalation will assist in reducing the likelihood of, and enable a period of monitoring for, any potential early adverse events. To further monitor safety due to the pharmacokinetics of CBD, participants will be referred for standard blood tests (liver and kidney function tests) by the trial nurse practitioner three times over the course of the trial (before enrollment, midpoint, post-treatment) to ensure no changes in hepatic or renal function.
The trial nurse practitioner will monitor all blood test results at each time point and communicate these to the participants' regular health professionals. Participants will not be enrolled in the trial if abnormal blood test results are received before baseline, and their participation will be discontinued if abnormal results are received at the midpoint. Participants will be encouraged to follow-up abnormal blood test results with their regular health professional at any time they are received. Referrals to medical practitioners and mental health counseling will be provided as appropriate.
In line with the Common Terminology Criteria for Adverse Events (CTCAE, v5), 55 in the event of a Grade 1 (mild symptoms without intervention) or Grade 2 (moderate symptoms with local or noninvasive intervention) adverse event, if the participant wishes to continue with the study following alleviated symptoms and renewed consent, they will be asked to refrain from administering their capsules for 24 h to allow time for recovery. In the event of a Grade 3 or above (medically significant but not life threatening or life-threatening consequences requiring urgent intervention) serious adverse event that is determined to be potentially related to consuming CBD, participation in the study will be terminated. In the event of a serious adverse event and unblinding of a participant is required for medical care, this will be performed by one of the senior investigators not directly involved in data collection who is unblinded to treatment allocations.
Participants may withdraw from the trial at any time but will undergo telephone monitoring for 3 weeks to ensure no withdrawal effects from the treatment (none anticipated). Any serious adverse events will be reported to the local Ethics Committee as required, and the decision to terminate the trial will be the responsibility of the chief investigator in collaboration with the trial senior clinicians.
Therapeutic trials of CBD in various clinical populations have utilized doses in the range of 200–1800 mg/day, with the most beneficial and therapeutic effects often observed at 300–600 mg.56–58 A dose of 300 mg/day is consistent with current trials investigating CBD as a treatment for major depression, anxiety, and Parkinson's disease.59–61 Furthermore, few and mild adverse events have been reported at 200–1800 mg/day for several weeks' duration,29,62–66 supporting the safety and tolerability of a 300 mg daily dose.
Procedure
Prebaseline and screening
The full testing schedule is outlined in Figure 1. Potential participants will complete a face-to-face prescreening interview, where the study inclusion and exclusion criteria will be assessed and the Rowland Universal Dementia Assessment Scale (RUDAS) 67 administered as a secondary measure of dementia symptom severity. Eligibility will then be confirmed with treating clinicians so that the trial is incorporated into patient care plans. The trial nurse practitioner will then refer participants for a standard blood test to assess hepatic and renal function to ensure no measurements of concern before participation.
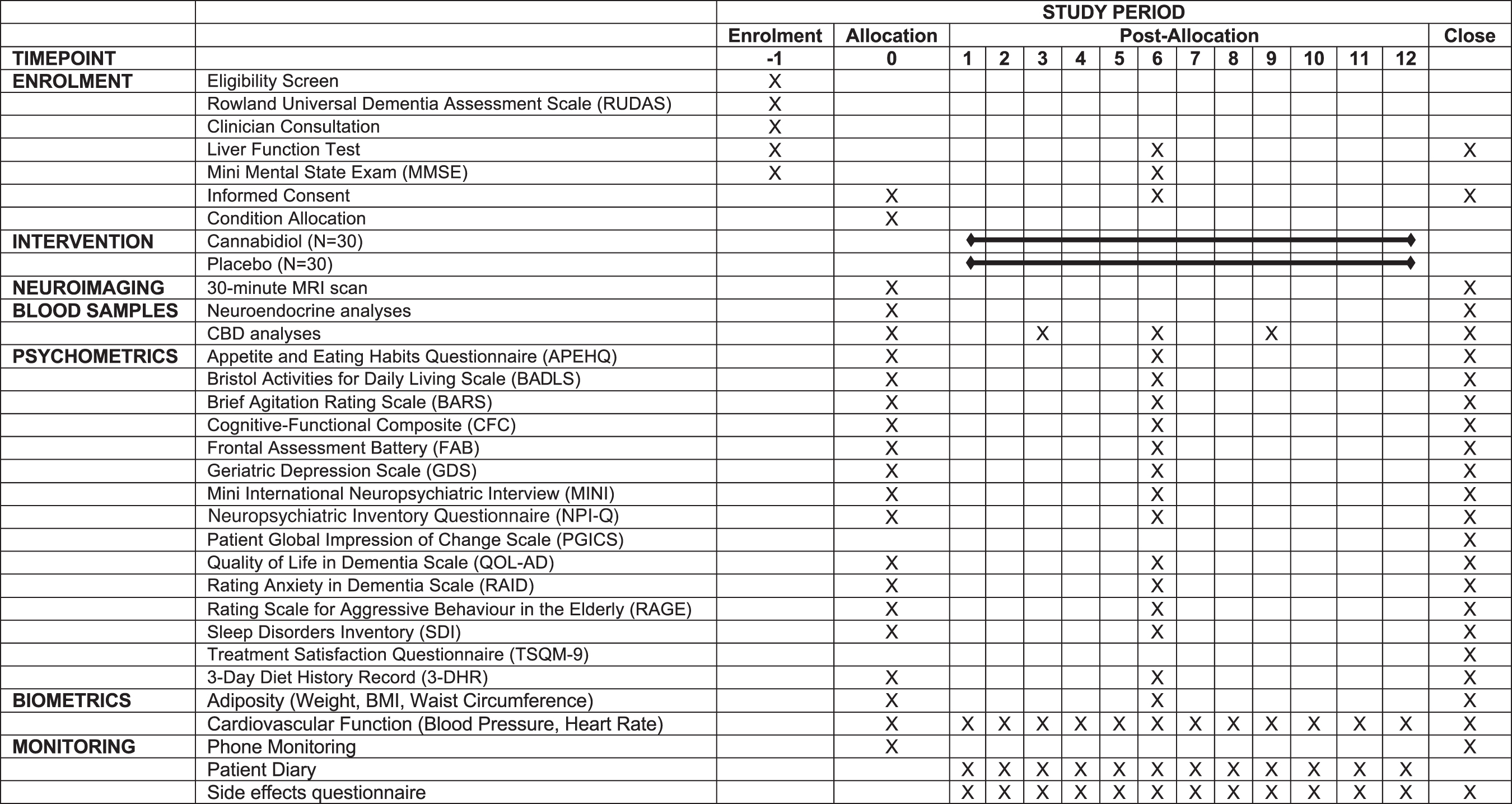
Testing schedule.
Baseline assessment
Blood sampling and psychometric assessment
The first day of baseline testing will be scheduled in the morning at the university clinical research trials unit for ∼2.5 h. For consistency in cognitive testing and to control for potential variations in blood-based neuroendocrine profiles, appointments will be scheduled between 9:00 and 11:30 am. On arrival, the study and testing procedures will be explained and written informed consent obtained from both the participant and their supporting person. Biometric measurements and a 10 mL nonfasting blood sample for neuroendocrine analyses will then be collected by the nurse. Neuroendocrine profiles will be quantified using ELISAs.
A comprehensive battery of validated clinician-administered and supporting person questionnaires will then be completed to assess cognitive, behavioral, and psychological symptoms of dementia. These will determine changes in cognition (CFC, 34 FAB 35 ); affect (GDS, 36 Mini International Neuropsychiatric Interview [MINI], 68 RAID 37 ); neuropsychiatric symptoms (Neuropsychiatric Inventory Questionnaire [NPI-Q] 40 ); appetite (APEHQ, 41 3-Day DHR); sleep (SDI 42 ); aggression and nonaggressive agitation (BARS 42 ; RAGE 43 ); and quality of life (QOL-AD, 38 BADLS 39 ).
Magnetic resonance imaging
The second day of baseline testing will take place at the local diagnostic imaging clinic and last ∼1 h. MRI scans will be completed the day after the baseline psychometric and neuroendocrine assessments (or at least within 1 week) and involve a 30-min scanning session using a 3.0T GE Architect scanner with a 48-channel head coil. Within each test session, three separate whole-brain three-dimensional (3D) T1-weighted images will be acquired using 3D inversion-prepared fast-spoiled gradient-recalled echo sequence (BRAVO): time echo/repetition time (TE/TR)=3.36 msec/8.59 msec, inversion time (TI)=450 msec, flip angle=12°, ARC=2, field of view (FOV)=25 cm, matrix 256×256×200, 1 mm3 isotropic voxels. The three T1 images will be upsampled to 0.6 mm3 isotropic, and aligned and averaged using MRTrix nonlinear alignment, to provide better structural contrast and detail for optimal segmentation of brain structures and subvolume measurements.
High angular resolution diffusion imaging (HARDI) diffusion-weighted imaging will be acquired using 147 directions, 3 b-values (b=700, 1000, and 2500, sec/mm2), and 8 b0 images at 2.5 mm3 isotropic resolution TR=4500 msec, TE=100 msec, and 3×multiband. A reverse phase-encoded gradient echo-planar imaging (EPI) sequence will be used to correct for distortion. These scans probe microstructural content of white matter and tracts to and from the brain regions of interest.
Drug dispensing and compliance
Weekly doses will be packaged into participant ID-labeled pharmacy-grade plastic containers and provided to participants and their supporting persons by the trial nurse. Participants will be provided with one labeled bottle each for their morning and evening doses, with two bottles used from the start of the trial in preparation for dose escalation. Participants will administer one 100 mg capsule twice daily, morning and evening (total 200 mg/day) for the first 2 weeks, where capsule administration will commence the evening after the completion of baseline assessments. Provided no serious adverse events (Grade 3 or above) were reported in the first 2 weeks, the evening dose will be escalated to 200 mg in the third week of the trial, for a total 300 mg daily dose for the remaining 10 weeks. Participants who report a serious adverse event (Grade 3 or above) before dose escalation will have their participation in the trial terminated.
Oral administration of capsules is consistent with standard medication-taking procedures and is therefore likely to increase adherence. Compliance with the capsule regimen will be informed by supporting persons, who will be requested to oversee and record capsule taking daily. Supporting persons will be further asked to return the old containers, including missed capsules, each week when the new supply is issued. Furthermore, plasma CBD metabolite concentrations in participants will be measured five times over the course of the trial (baseline, week 3, midpoint, week 9, and post-treatment) to monitor the level of cannabinoid metabolites and corroborate self-report compliance with the treatment regime.
Telephone contact will be maintained with participants for the first 3 days of initial capsule administration following the baseline assessment, and for 2 days following dose escalation, to monitor potential adverse events. Telephone calls for monitoring are expected to last no longer than 10 min duration. During such phone calls, participants will be asked to free recall how they have been feeling since starting their capsules and supporting persons will be asked to free recall if they have noticed any changes in participant behavior, health, or psychological functioning.
Weekly home monitoring
Brief (30-min) weekly home visits will be conducted with the trial nurse, participant, and supporting person for the purposes of monitoring of participant health, engagement with the trial, and weekly supply of capsules. During these visits, cardiovascular health (heart rate, blood pressure) will be monitored by the trial nurse to ensure no changes in functioning. Weekly patient diaries and a brief questionnaire will be used to monitor the presence and severity of any short and long-term adverse events that may relate to CBD or placebo treatment. Weekly patient diaries will be provided to participants and their supporting person to complete over the course of the week between home visits, consisting of a grid where patient reported or supporting person-reported adverse events can be recorded such as gastrointestinal distress, fatigue, drowsiness, or headaches, their severity or change in severity or frequency, and any medical consultations or remedies undertaken.
The questionnaire requires participants to reflect on the previous week and rate the severity of a list of symptoms that may be related to CBD or placebo intake, such as gastrointestinal distress, fatigue, drowsiness, or headaches, on a 4-point Likert scale of “Not at All” to “Severe,” with space provided for either participants or supporting persons to note any additional information. Participants will be directed to their regular health professional if any symptoms or measurements of concern in these parameters are reported as a new symptom or increase in severity or frequency as compared with before the trial. In the third and ninth weeks, participants will be asked to refrain from taking their morning capsule until a 5 mL blood sample is obtained by the trial nurse to monitor CBD levels.
Midpoint assessment
At the midpoint (6 weeks) of the trial, participants and their supporting person will attend the university for ∼1 h. This session will include briefer versions of baseline assessments, a subset of cognitive tests, biometric measurements, and a 10 mL blood sample (neuroendocrine/CBD). Participants will be provided with their weekly supply of capsules. Participants will also be referred for a standard blood test by the trial nurse practitioner to ensure no changes in hepatic or renal function since the start of participation. If any significant deterioration from baseline is detected by the trial nurse practitioner, who will communicate results as appropriate to the participants' treating clinician (e.g., in liver function), the participant will be discontinued from the trial.
Post-treatment assessment
At the conclusion of the 12 weeks, participants and their supporting person will complete a post-treatment session replicating baseline format. On the first day, biometric measures and a 10 mL blood sample will be obtained and questionnaires completed. Participants will administer the final CBD dose the night before their MRI (12 h washout). If the MRI is not scheduled the following day, participants will be asked to maintain capsule administration until the night before. Following the MRI, participants will complete the Treatment Satisfaction Questionnaire for Medication (TSQ-M) 69 and the Patient Global Impression of Change Scale (PGICS) 70 to index perceived post-treatment change. Subsequently, participants and supporting persons will be advised by letter from the research team disclosing their treatment condition. Telephone contact will be maintained with participants to monitor potential withdrawal effects (none anticipated); daily in the 2 days following the MRI, twice/week for the following week, and then once/week for 2 weeks.
Privacy and data management
Participant and supporting person privacy and confidentiality will be maintained at all times. All data collected will be deidentified and coded with an ID number only, with participant or supporting person names and contact information written on consent forms only. All hard copy data, including personal information, will be stored in a locked filing cabinet in a secure location accessible by the trial coordinator during data collection, and for 15 years after the trial has concluded. Electronic data will be securely stored on password-protected share drives. Biological samples will be stored in a locked and secure −80° freezer.
A data safety management board will not be implemented for this trial due to the small sample size. The research team will be responsible for participant recruitment and monitoring, the development of trial resources such as information sheets, decisions to amend the study protocol, coordination of the trial, data quality control, and the development of a trial database. Data will be stored on a secure server managed by the UOW and coded using SPSS (version 28) and JASP (version 0.16.2). Missing data will be handled using multiple imputation where applicable and data screening will be conducted by investigating ranges to identify impossible scores. Trial outcomes will refer to aggregate data only, and no individual or identifying information will be released. Aggregate data will be reported in peer-reviewed academic publications and academic conference presentations.
Ethics guidelines
This trial has received full approval from the Joint University of Wollongong and Illawarra Shoalhaven Local Health District Health and Medical Ethics Committee (2020/ETH02708). The trial will be conducted in accordance with the Australian National Statement on Ethical Conduct in Human Research (2007), as revised in 2018, prepared by the National Health and Medical Research Council (NHMRC). The trial is also registered with the Australian Therapeutic Goods Administration (CT-2020-CTN-03849-1v2) and the ANZCTR (ACTRN12621001364864).
Outcome measures and analysis
Neuroanatomical changes will be measured at baseline and post-treatment. T1-weighted scans will be used to index volumes of cortical and subcortical brain regions, including the hippocampus, and tractography will be used to measure changes in white matter connectivity and neural growth. Changes in neuroendocrine profiles will also be assessed at baseline and post-treatment. Peripheral neuropeptide BDNF will index neural growth, differentiation, and maturation. Proinflammatory cytokines TNF-α and interleukin-6, proinflammatory adipokine leptin, stress hormone cortisol, and proinflammatory and oxidative stress marker plasma serotonin will be used as indices of inflammation and oxidative stress. Cognitive, behavioral, and psychological profiles, including cognition, affect, aggression and nonaggressive agitation, psychosis, appetite and eating behaviors, sleep patterns, and quality of life, will be assessed at baseline, at the midpoint of the study, and post-treatment. Adiposity, as indexed by BMI and waist circumference, and cardiovascular function, indexed by blood pressure and heart rate, will be assessed at each primary time point, the latter also during the weekly home visits.
Statistical analyses
Statistical analyses will be conducted using SPSS (Version 27). Mixed model repeated measures analyses of variance (rmANOVAs) will be used to test for differences in the outcome measures, with the between-subjects factor of treatment (CBD, placebo) and within-subjects factor of time (baseline, midpoint where applicable, post-treatment). Covariates will include, where appropriate, age, sex, years of education, and dietary fat intake. Pearson's correlations will be used to determine relationships between variables. Missing data will be handled using multiple imputation and data screening will investigate ranges to identify impossible scores. For all analyses, α < 0.05 will be considered statistically significant. Post hoc analyses will be conducted using Bonferroni corrections. If data are found to violate the assumptions of the statistical tests, nonparametric analyses will be used. Results from the study will be disseminated in aggregate format in academic journals, academic conference presentations, and may be reported in general media.
Power calculations
There are few studies that can be used to estimate sample size for this trial, as the proposed protocol is among the first to investigate the efficacy of isolated oral CBD in improving neuroanatomical and neuroendocrine changes, in addition to behavioral and psychological symptoms, of early-stage dementia. Our previous trial administering 200 mg daily doses of isolated oral CBD in 20 chronic cannabis users, a population also vulnerable to hippocampal changes and psychopathology, represents the most comparable study on which to base sample size calculations. Results from our previous trial indicate medium effect sizes for improvement in the primary outcomes of hippocampal subfield volumes (d=0.63), cognition (d=0.54), depression (d=0.39), and anxiety (d=0.38).18,29
Consequently, for N=60, representing the target sample size, G*Power (version 3.1.9.4) calculations indicate that with α=0.05, there is ∼90% power to detect a medium effect size (d=0.50) for the hypothesized changes to brain structural integrity, neuroendocrine profiles, cognition, and behavioral and psychological symptoms of dementia (BPSD; F-test analysis of variance repeated measures, within-between interaction, two groups), and ∼80% power to detect a medium effect size bivariate correlation of 0.45 between measures. For N=40, accounting for a potential attrition rate of 33% at follow-up, there is 85% power to detect a medium effect size for changes in the outcome measures of interest (rmANOVA), and 75% power to detect a bivariate correlation of 0.45 between measures.
Discussion
Establishing new, effective, and accessible treatments to reduce the impacts of early-stage dementia and improve the prognosis and quality of life of affected individuals is a public health imperative. This trial is novel in proposing CBD as one such treatment, with accumulating evidence suggesting CBD may be a promising candidate to address multiple facets of dementia.
This study protocol has several identifiable strengths. The study design consists of three comprehensive assessment time points and weekly monitoring, with an extensive range of outcome measures that allows more thorough and combined neurobiological and symptom-level investigation of pure CBD treatment efficacy than in existing trials in early-stage dementia.30–33 The study also includes a detailed safety protocol designed to maximize participant safety, including the dose-escalation approach to CBD administration, weekly monitoring by a nurse, weekly assessment of potential adverse events, and detailed procedures in the event of any adverse reaction. Relatedly, the strict list of inclusion and exclusion criteria reduces the likelihood of potential confounding effects between CBD and pre-existing physical and psychological conditions, and medications, further ensuring participant safety and specificity of outcomes.
However, limitations to the proposed study should be acknowledged. The inclusion and exclusion criteria may impede participant recruitment and limit the generalizability of findings. Furthermore, the combined effects of age, diagnoses, potential frailty, and the intensive nature of the trial may result in high attrition.
To our knowledge, this study will be among the first to investigate isolated CBD in improving neurobiological and psychological outcomes in individuals living with early-stage dementia. If effectiveness of CBD is determined, these findings may support implementation of an alternative treatment that aims to reduce the risks and impacts of the disease, improve functioning, quality of life, and prognosis for affected individuals. Participant recruitment for the study will begin in May 2022 and is estimated to conclude in March 2023. Outcomes are expected in 2023 and 2024.
Footnotes
Authors' Contributions
All authors were involved in the study design. J.G.B. prepared the protocol, and all authors contributed to and approved the final version of the protocol.
Author Disclosure Statement
The authors declare no potential conflicts of interest.
Funding Information
Funding for the study was provided by the Australian Center for Cannabinoid Clinical and Research Excellence (ACRE), a National Health and Medical Research Council (NHMRC) Center for Research Excellence (CRE; APP1135054), the University of Wollongong (UOW), and the Illawarra Health and Medical Research Institute (IHMRI). L.-M.G. was supported by a National Institutes Grant (NIG) held by the Australian National University. No funding agencies had any further role in study design, in the writing of this protocol, or in the decision to submit the article for publication.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.