
Abstract
Introduction:
Cannabidiol (CBD) has important pharmacological activity, which includes antispasmodic, antioxidant, antithrombotic, and antianxiety properties. CBD has been applied as a health supplement to atherosclerosis. However, CBDs effect on gut microbiota and metabolic phenotype is unclear.
Materials and Methods:
We constructed a high production of cardiovascular risk factors, such as trimethylamine-N-oxide (TMAO) and phenylacetylglutamine (PAGln), in a mouse model using Clostridium sporogenes colonization. We used 16S ribosomal RNA (rRNA) gene sequencing and ultra-high performance liquid chromatography-quadrupole time-of flight mass spectrometry-based metabolomics to evaluate the effect of CBD on gut microbiota and plasma metabolites.
Results:
CBD decreased the levels of creatine kinase (CK), alanine transaminase (ALT), and low-density lipoprotein cholesterol and markedly increased high-density lipoprotein cholesterol. Furthermore, CBD treatment increased the abundance of beneficial bacteria, which include Lachnospiraceae_NK4A136 and Blautia in the gut, but it decreased the levels of TMAO and PAGln in the plasma.
Conclusion:
CBD might have beneficial effects for cardiovascular protection.
Introduction
In the past 10 years, it has become clear that gut microbiota play a crucial role in both human health and disease.1,2 Microbial transplantation studies allow for a clear transmission of a host phenotype or disease susceptibility, which has provided strong evidence in support of the causative role of gut microbiota in host physiological processes and disease risks. 2 It is well known that Clostridium sporogenes is important in the production of trimethylamine-N-oxide (TMAO) and phenylacetylglutamine (PAGln), which are metabolites derived from gut microbiota that have been identified as potent risk factors for cardiovascular diseases (CVDs).3,4 Therefore, in this study, we constructed a C. sporogenes colonization mouse model to explore the effects on the host gut microbiota and metabolic phenotype, especially on the gut microbiota-derived risk factors of TMAO and PAGln.
Cannabis is one of the oldest cultivated plants purported to have unique medicinal properties. There are more than 100 kinds of cannabinoids isolated from cannabis plants that have been identified. 5 Among them, the more abundant components are tetrahydrocannabinol (THC) and cannabidiol (CBD). 6 CBD has numerous biological actions, such as anti-inflammatory, anti-arthritic, antianxiety, antiproliferative, anticonvulsant, immunomodulatory, and neuroprotective properties, 7 but it is not intoxicating.8,9 Metabolomics reflects the metabolic profile of biological samples. In addition to reflecting diagnostic and predictive information on biochemical effects, metabolomics describes the changes in metabolic profiles in biological samples in detail.10–12 However, few studies have paid attention to the effects of the alteration of CBD-induced gut microbiota on host metabolism, especially using a microbial transplantation model.
In this study, 16S ribosomal RNA (rRNA) gene sequencing and the ultra-high performance liquid chromatography-quadrupole time-of flight mass spectrometry (UPLC/QTOF-MS)-based metabolomics were used to evaluate the effect of CBD on gut microbiota and plasma metabolites. The results showed that CBD increased the abundance of beneficial bacteria in the gut and decreased the levels of cardiovascular risk factors in the plasma. All our evidence indicated that CBD might offer beneficial effects for cardiovascular protection.
Materials and Methods
Reagents and materials
Ampicillin, vancomycin, metronidazole, and neomycin were purchased from Aladdin Chemical Reagent Co., Ltd. (Shanghai, China). CBD was purchased from Desite Technology Development Co., Ltd. (Chengdu, China). Fmoc_Glycine (internal standard substance) was purchased from Belling (Beijing, China). High-purity chemicals, which included formic acid and acetonitrile, were purchased from Thermo Fisher Scientific (Shanghai, China).
Ethical approval and treatment of mice
Animal protocol (JN. No. 20210915c1200230[308]) was approved by the Committee of Jiangnan University. All experiments were performed in compliance with the relevant laws and institutional guidelines. Female C57BL/6J mice (8 weeks old) that were purchased from Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China) were kept in the animal facility at Jiangnan University. Mice were assigned randomly to two groups (n=5 per group) and given 1 week to acclimatize to the environment. Antibiotics were received for 2 weeks to induce gut dysbiosis, using a previously established pharmacological model.13,14 After clearing the intestinal microbiota, all mice were gavaged with C. sporogenes for 2 weeks. After colonization, CBD was administered to mice (CBD group) at a daily dose of 50 mg/kg of body weight dissolved in olive oil, and the other group was fed olive oil (C. sporogenes group). After 4 weeks of gavage with CBD, all mice were sacrificed, and plasma and feces were collected for further analysis.
16S rRNA sequencing analysis of fecal bacteria
The bacterial DNA from mouse feces was extracted using a genomic DNA kit (Tiangen Biotech, Beijing, China) according to the manufacturer's instructions. Gel electrophoresis was used to evaluate the DNA sample quality, and NanoDrop was used to determine the concentration. The V3-V4 region of the bacterial 16S rRNA genes was amplified from the entire genome using the primers (515F [5′-GTGCCAGCMGCCGCGGTAA-3′] and 806R [5′-GGACTACHVGGGTWTCTAAT-3′]). An Illumina HiSeq platform was used to run the high-throughput sequencing on the amplicon libraries. Using Usearch 15 and the Greengenes Database, 16 the acquired sequences were clustered into operational taxonomic units based on 97% similarity. Statistical analysis was conducted using R and bioinformatics analysis.
Extraction of plasma and fecal metabolites
Plasma protein was precipitated by adding four volumes of ice-cold methanol (with an internal standard) with vortex mixing for 30 sec. After centrifugation (14,000 rpm, 10 min, 4°C), the supernatant was collected for UPLC/QTOF-MS analysis.
Fecal samples were thawed and weighed. Three milligrams of fecal samples combined with 200 μL of 50% ice-cold methanol (with an internal standard) were prepared to extract fecal metabolites, vortexed, and extracted ultrasonically in an ice bath (30 min). After centrifugation (14,000 rpm, 10 min, 4°C), the supernatant was collected for UPLC/QTOF-MS analysis.
UPLC/QTOF-MS analysis
Chromatographic conditions: Chromatographic analysis was performed on UPLC (AB SCIEX ExionLC) and TOF-MS (AB SCIEX, Triple TOF 5600+). Plasma samples were analyzed using a Kinetex C18 column (100×2.1 mm, 2.6 μm; Phenomenex) and a BEH amide column (100×2.1 mm, 1.7 μm; Waters). Fecal samples were analyzed using a Kinetex C18 column (100×2.1 mm, 2.6 μm; Phenomenex) and a BEH Z-Hilic (100×2.1 mm, 1.7 μm; Thermo Scientific). During C18 column separation, the ingredients of the mobile phase were (A) water and (B) acetonitrile, and each contained 0.1% formic acid.
The elution program was as follows: 0–1 min, 5% B; 1–11 min, 5–95% B; 11–13 min, 95% B; 13–14 min, 95–5% B; 14–15 min, 5% B. During BEH column separation, the mobile phase was composed of solvent A (10 mM ammonium formate in H2O) and solvent B (10 mM ammonium formate in 90% acetonitrile/10% H2O). The elution program for plasma samples was as follows: 0–1 min, 100% B; 1–11 min, 100% B to 70% B; 11–12.5 min, 70% B; 12.5–13 min, 70% B to 100% B; 13–15 min, washing with 100% B. The elution program for fecal samples was as follows: 0–3 min, 95% B; 3–11 min, 95% B to 60% B; 11–12.5 min, 60% B to 95% B; 13–15 min, washing with 95% B. The flow rate was 0.3 mL/min, column temperature was 40°C, and injection volume was 2 μL.
Mass spectrometry conditions included electrospray ionization source, ion source voltage was set at 5500 and −4500 V in the positive and negative modes, respectively, gas temperature was 350°C, gas flow was 12 L/min, atomizer nitrogen flow was 30 psi, source temperature was 550°C, and collision energy was 35±15 V. Data were collected in centroid mode from 100 to 1000 m/z for fecal samples and from 50 to 1000 m/z for plasma samples.
Data processing
Metabolic findings were gathered as *.wiff files, which MSConvert then transformed into *. mzXML files. All MS data were compiled, aligned, and normalized, and peak tables were created using MZmine 2.53 and Microsoft Excel 2020. The peak table was then uploaded to MetDNA for metabolite identification. 17 The processed data list was then imported into SIMCA-P (version 14.1) for orthogonal partial least squares-discriminant analysis (OPLS-DA). The parameters R2X and Q2Y were stable and good for fitness and prediction. The difference metabolites were screened according to the fold change (FC) against control samples. Only features with FC >2.0 and FC > 0.5, p<0.05, were considered further. We used R to draw a heat map to visualize the relative content of different metabolites in each group.
A possible relationship between the altered plasma, fecal metabolites, and the genus-level of gut microbiota was investigated using Spearman's correlation analysis. We used R to draw a correlation heat map to visualize the significant correlation values between gut microbiota genera and altered metabolites.
Statistical analysis
All data are presented as mean±standard error, and unpaired t-tests on the data from two groups were utilized for statistical analysis. The error bars, commonly known as *p<0.05, **p<0.01, ***p<0.001, represent the mean and standard error. SPSS 16.0 and GraphPad Prism 8.0 were used to analyze the data.
Results
Evaluation of C. sporogenes colonization model
A common antibiotic combination of ampicillin, neomycin sulfate, metronidazole, and vancomycin was able to clear the intestinal microbiota. After clearing the intestinal microbiota, we introduced C. sporogenes into the gut to build a high production of cardiovascular risk factors in the mouse model, such as TMAO and PAGln. There were significant (p<0.05) variations in the alpha diversity (Fig. 1a) and beta diversity (Fig. 1b, c), which demonstrated that C. sporogenes colonization altered the variety of the intestinal microbiome significantly. In comparison to the antibiotic treatment group (Abx), the C. sporogenes group increased the relative abundance of Clostridium at the genus level significantly (Fig. 1d). The increase in plasma TMAO and PAGln levels in the C. sporogenes colonization group also indicated that C. sporogenes colonized successfully (Fig. 1e).
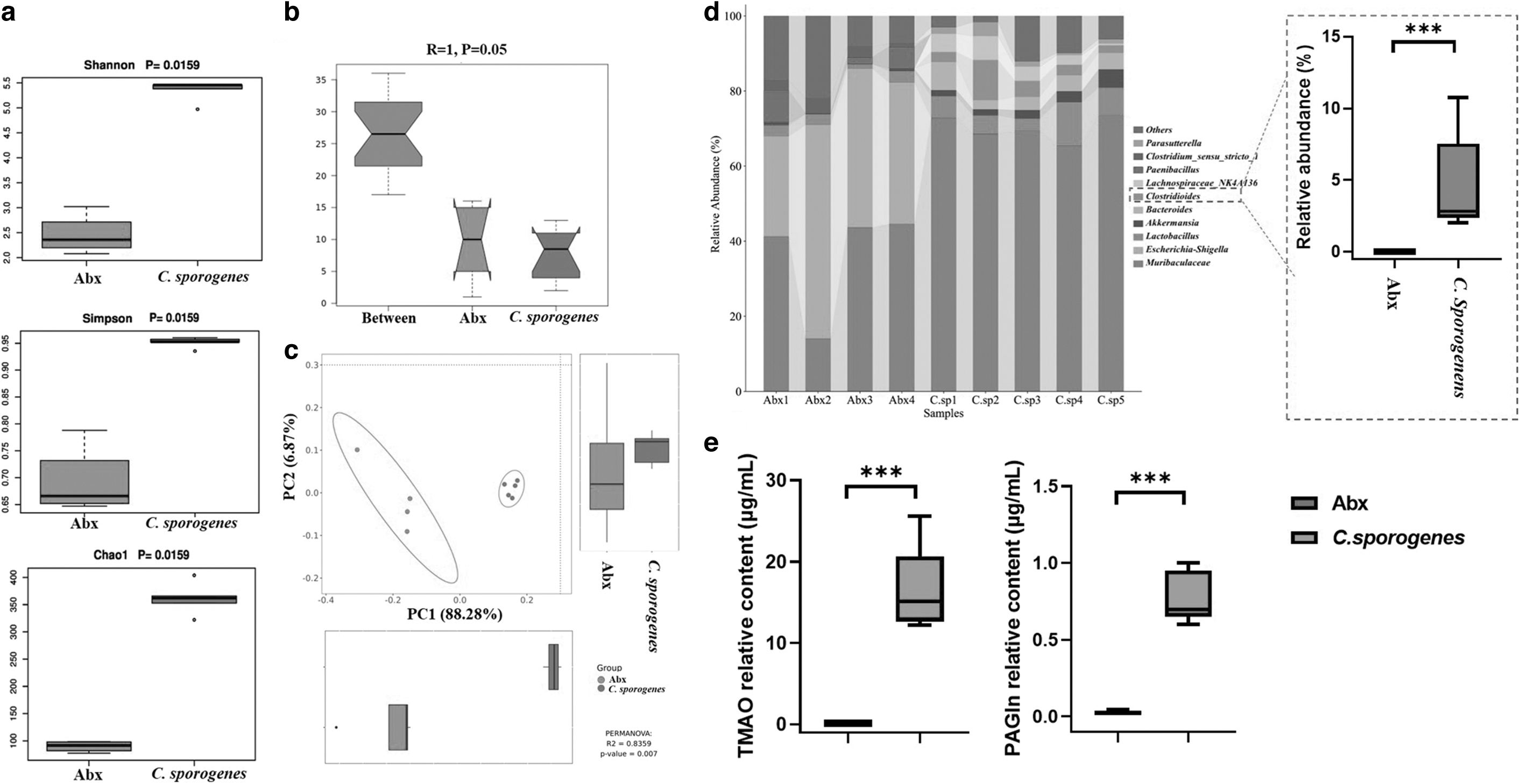
Evaluation of Clostridium sporogenes colonization model.
Effect of CBD on biochemical parameters
During CBD treatment, there was no difference in body weight between the CBD group and the C. sporogenes group, although the food intake of the CBD group was higher than that of the C. sporogenes group (Fig. 2a). We also analyzed the differences in the biochemical parameters. We did not find any differences in levels of lactic dehydrogenase (LDH), aspartate transaminase (AST), total bile acids (TBA), creatine, triglycerides (TG), or total cholesterol (TC). We found that the levels of creatine kinase (CK), alanine transaminase (ALT), and low-density lipoprotein cholesterol (LDL-C) were significantly lower in the CBD group, and high-density lipoprotein cholesterol (HDL-C) was significantly higher in the CBD group (Fig. 2b–d).
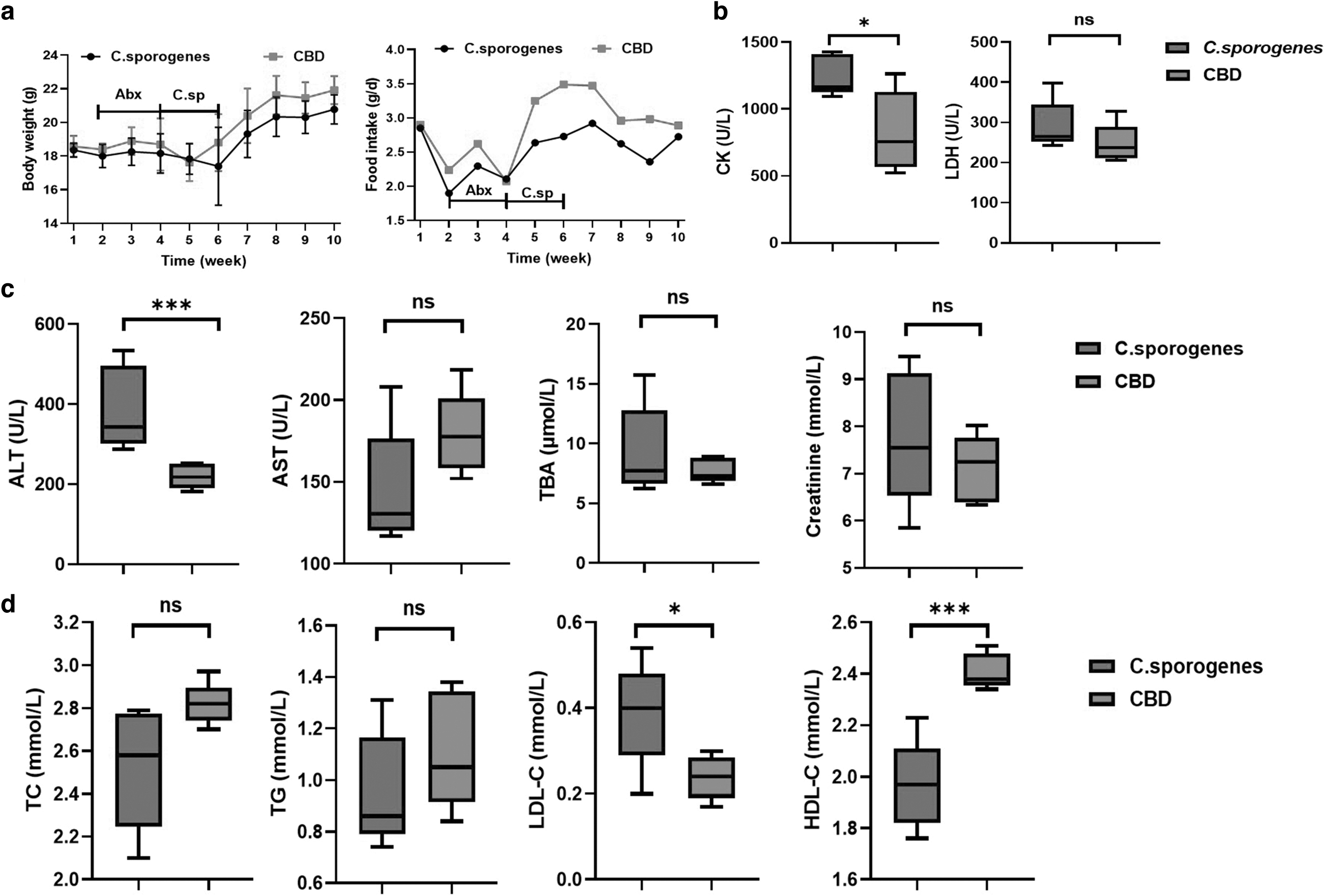
Effects of CBD on biochemical parameters.
Effect of CBD on composition of gut microbiota
We used 16S rRNA gene sequencing to determine the composition of microbiota. There were no significant variations in the alpha diversity indices (Chao1, Shannon, or Simpson indices) between the C. sporogenes group and the CBD group (Fig. 3a). The beta diversity refers to the variations in species composition on a spatial scale, and it used to evaluate group resemblance. The intestinal microbial structure of the C. sporogenes and CBD groups was different (Fig. 3b), which demonstrated that CBD altered the diversity of species in the gut microbiome. The principal component analysis (PCoA) scores clearly distinguished the community composition between the C. sporogenes group and the CBD group (Fig. 3c).

Effect of CBD on microbiota composition in mice.
Moreover, Bacteroides, Proteobacteria, Firmicutes, and Verrucomicrobia made up almost 99% of the total amount of bacteria. Firmicutes and Desulfobacterota levels increased significantly after CBD treatment, but Bacteroidota levels decreased (Fig. 3d, e). At the genus level, we discovered 10 different genera between the C. sporogenes group and the CBD group (Fig. 3f). Comparison with the C. sporogenes group, CBD treatment increased the relative abundance of Lachnospiraceae_NK4A136, Desulfovibrio, Blautia, Turicibacter, and GCA-900066575 significantly, but the treatment decreased the relative abundance of Muribaculaceae, Bacteroides, Alistipes, and Muribaculum significantly (Fig. 3g). These findings suggested that compared with the C. sporogenes group, the CBD group had significant alterations in the gut microbiota at the phylum and genus levels.
Effect of CBD on fecal metabolic profiles
The metabolic profiles of the C. sporogenes and CBD groups were compared using a supervised multivariable statistical technique called OPLS-DA, which revealed that the metabolic profile of the CBD group was distinct from the C. sporogenes group (Fig. 4a). The model parameters R2X, R2Y, and Q2 were all >0.5, which is the threshold value for an acceptable predictive model in metabolomics analysis. The fecal metabolic profile of the CBD group differed from that of the C. sporogenes group. The OPLS-DA model's permutation test diagram further demonstrated the model's robustness and ability to discriminate samples (Fig. 4b). Volcano plots were used to identify different metabolites with FC >2 and FC <0.5, p-value <0.05; as a result, there were 39 altered metabolites (Fig. 4c). The heat map described the affected metabolites (Fig. 4d).
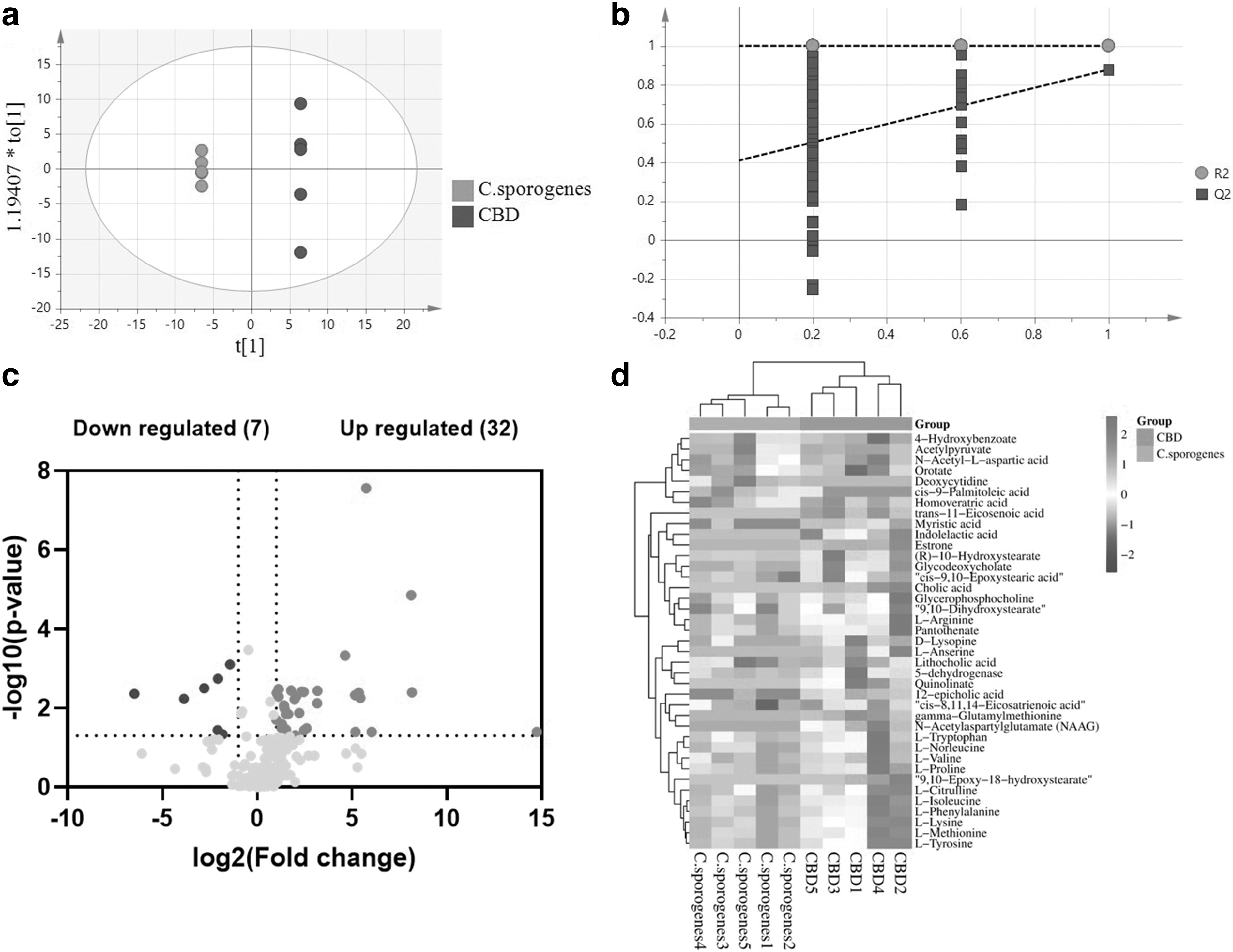
Effect of CBD on metabolite profile of fecal samples.
Among these metabolites, 4-hydroxybenzoate, acetylpyruvate, n-acetyl-L-aspartic acid, orotate, deoxycytidine, cis-9-palmitoleic acid, and homoveratric acid decreased in the CBD group, and L-valine, cis-8,11,14-eicosatrienoic acid, cis-9,10-epoxystearic acid, lithocholic acid (LCA), glycerophosphocholine, L-citrulline, pantothenate, L-norleucine, L-isoleucine, L-tryptophan, L-phenylalanine, L-lysine, L-tyrosine, 9,10-dihydroxystearate, 12-epicholic acid, L-methionine, myristic acid, 5-dehydrogenase, L-proline, L-arginine, (R)-10-hydroxystearate, D-lysopine, glycodeoxycholate, L-anserine, trans-11-eicosenoic acid, gamma-glutamylmethionine, indolelactic acid, N-acetylaspartylglutamate (NAAG), 9,10-epoxy-18-hydroxystearate, quinolinate, estrone, and cholic acid increased significantly.
Effect of CBD on plasma metabolic profiles
The OPLS-DA model was used to evaluate the metabolic profiles of plasma samples between the C. sporogenes group and the CBD group (Fig. 5a, b). The metabolic profile of the CBD group differed from that of the C. sporogenes group. The volcano plots were used to identify 13 altered metabolites with FC >2 and FC <0.5, p-value <0.05 (Fig. 5c). The heat map described the affected metabolites treated with CBD (Fig. 5d). Among these metabolites, cirsimaritin, angelicin, PAGln, 2-oxindole-3-acetic acid, TMAO, n-benzoyl glycine, and n-acetylphenylalanine decreased in the CBD group, and hexanoyl glycine, chaulmoogric acid, LPC18:3, brevicarine, 1-monostearin, and palmitoylcarnitine increased.
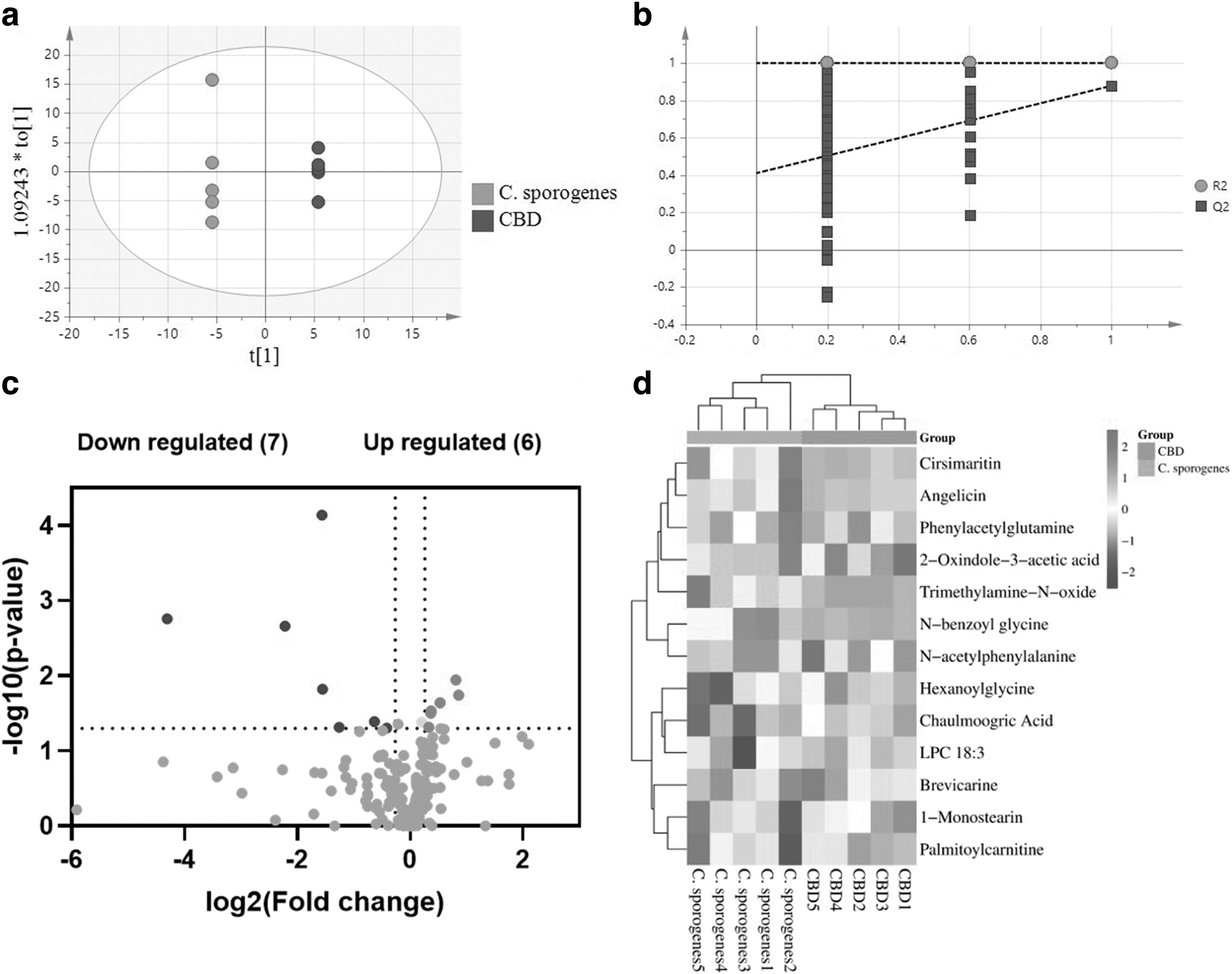
Effect of CBD on metabolite profile of plasma samples.
Correlations between gut microbiota and altered metabolites
The Pearson correlation analysis was utilized to find possible connections between gut microbiota genera and altered metabolites. 4-Hydroxybenzoate and n-acetyl-L-aspartic acid were correlated with all affected gut microbiota genera (Fig. 6a). NAAG, D-lysopine, and gamma-glutamylmethionine were correlated positively with Lachnospiraceae_NK4A136, GCA-900066575, Blautia, and Desulfovibrio and correlated negatively with Alistipes, Bacteroides, Muribaculum, and Muribaculaceae. Amino acids and their derivatives, such as L-lysine, L-isoleucine, L-phenylalanine, L-tryptophan, L-tyrosine, L-methionine, L-proline, and D-lysopine, were all correlated positively with Blautia and Desulfovibrio and correlated negatively with Muribaculum and Muribaculaceae. 9,10-Dihydroxystearate, estrone, cis-9,10-epoxystearic acid, glycodeoxycholate, cis-8,11,14-eicosatrienoic acid, and myristic acid were correlated positively with Blautia and Turicibacter and correlated negatively with Alistipes, Bacteroides, and Muribaculum.
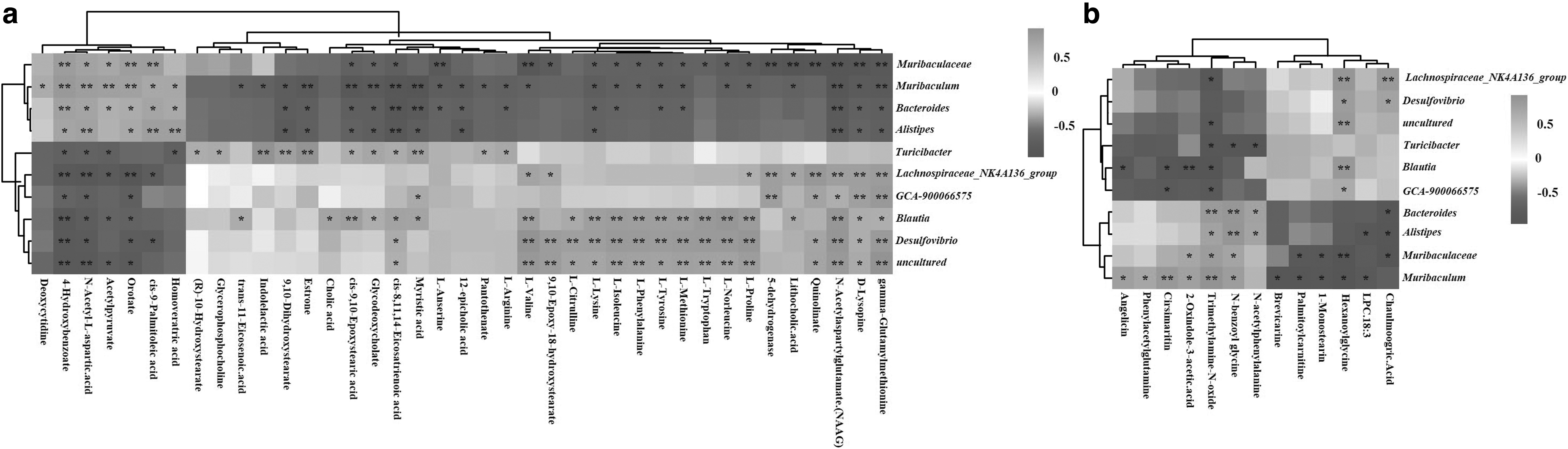
A correlation heat map of the altered gut microbiota genera and (a) altered fecal and (b) plasma metabolites. *p < 0.05, **p < 0.01.
The plasma metabolites, cirsimaritin, 2-oxindole-3-acetic acid, and TMAO were correlated positively with Muribaculum and correlated negatively with Blautia. TMAO, 2-oxindole-3-acetic acid, and n-benzoyl-glycine were correlated positively with Muribaculaceae and Muribaculum. TMAO, n-benzoyl-glycine, and n-acetylphenylalanine correlated positively with Alistipes and Bacteroides and correlated negatively with Turicibacter. Palmitoylcarnitine, 1-monostearin, and hexanoylglycine were correlated negatively with Muribaculaceae and Muribaculum (Fig. 6b).
Metabolic pathway related to altered gut microbiota
MetaboAnalyst 5.0 (www.metaboanalyst.ca) was used to map the relevant pathways identified by statistical correlations. As a result, there were six altered metabolic pathways implicated in CBD-induced changes in the gut microbiota and plasma metabolites (p<0.05). These six pathways included aminoacyl-tRNA (transfer RNA) biosynthesis, phenylalanine metabolism, phenylalanine, tyrosine, and tryptophan biosynthesis, valine, leucine, and isoleucine biosynthesis, arginine biosynthesis, and pantothenate and coenzyme A (CoA) biosynthesis (Fig. 7).

Summary of pathway analysis.
Discussion
Healthy gut microbiota is necessary for normal metabolic processes and microecological balance, and changes in the composition of gut microbiome may lead to obesity, diabetes, and other conditions by altering the metabolism of the host.18,19 Compared with the C. sporogenes group, the relative abundance of Bacteroidetes in the CBD group decreased, but Firmicutes and Desulfobacterota increased, which demonstrated that CBD boosted Firmicutes and Desulfobacterota and inhibited Bacteroidetes markedly. A high-fiber diet increased the abundance of Firmicutes and reduced the abundance of Bacteroides, which consequently increased the concentration of short-chain fatty acids (SCFAs) in the intestine. 20
CBD administration resulted in a significant increase in the relative abundance of Lachnospiraceae_NK4A136, Desulfovibrio, Blautia, Turicibacter, and GCA-900066575 but decreased the relative abundance of Muribaculaceae, Bacteroides, Alistipes, and Muribaculum significantly. Lachnospiraceae is the main producer of SCFAs. The butyrate-producing bacteria Lachnospiraceae_NK4A136, Blautia, and Turicibacter were associated with improved gut barrier function and decreased weight gain.21–23 Butyrate has heart-healthy properties and may help lower cholesterol levels, and reduce the risk of developing heart disease. Butyrate also helped to produce gut hormones that regulated blood sugar level. 24 Muribaculum is a member of the Bacteroidetes phylum.
With an associated mortality of more than 19%, Bacteroides species are significant clinical pathogens that were present in most anaerobic infections. 25 Alistipes, which is a relatively new genus of bacteria discovered in medical clinical samples, was linked to mental disorders, colorectal cancer, liver fibrosis, and CVD. 26 The number of potential beneficial bacteria increased as the number of potential pathogenic bacteria decreased, and because of this, we hypothesize that CBD regulated host metabolism.
Microbial metabolites are an important factor in the study of host–microbial interaction. Microbiota-derived metabolites, notably tryptophan and indole derivatives, TMAO, SCFAs, bile acids (BAs), and branched-chain amino acids (BCAAs), are related to the pathogenesis of metabolic disorders. 27 After CBD treatment, fecal metabolites of indole-3-lactic acid (ILA), 9,10-dihydroxystearic acid, estrone, LCA, glycerophosphocholine, and L-citrulline increased significantly. ILA, which is tryptophan's metabolite, is an anti-inflammatory substance. 28 Elevated ILA was positively related to Turicibacter and negatively related to Muribaculum. 9,10-Dihydroxystearic acid is an oxidation product of oleic acid, which improved glucose tolerance and insulin sensitivity. 29 Estrone promoted the production of nitric oxide, which helped to build healthier blood vessels and improved heart function. 30
In this finding, we found that 9,10-dihydroxystearic acid and estrone were related positively to Turicibacter and related negatively to Alistipes, Bacteroides, and Muribaculum. LCA is a monohydroxy BA produced by gut microbiota that has antitumor and anti-inflammatory properties. 31 Elevated LCA was associated with increased Lachnospiraceae_NK4A136 and Blautia and decreased Muribaculaceae and Muribaculum. Glycerophosphocholine is a natural choline that can be metabolized by gut microbiota to produce TMAO, which was related to high CVD risk.32–34 Increased Turicibacter was linked to elevated glycerophosphocholine. L-citrulline had positive effects on atherosclerosis, insulin resistance, inflammation, and hypertension. 35 Blautia and Desulfovibrio were positively related to L-citrulline levels.
After CBD treatment, plasma metabolites of cirsimaritin, angelicin, PAGln, 2-oxindole-3-acetic acid, TMAO, n-benzoyl-glycine, and n-acetyl-phenylalanine decreased. TMAO and PAGln, which are both metabolic products of gut microbiota, have been associated with an increased risk of CVDs, diabetes, and cancer.4,36 CBD treatments promote the growth of beneficial bacteria and reduce the microbiota-derived metabolites TMAO and PAGln. We discovered that TMAO levels were positively related to Muribaculum, Muribaculaceae, Alistipes, and Bacteroides and negatively related to Lachnospiraceae_NK4A136, Turicibacter, and Blautia. Muribaculum was positively related to PAGln levels. Previous studies indicated that C. sporogenes increased the levels of TMA, TMAO, and PAGln in the host.2,37 Similarly, we confirmed that CBD reversed the increase in TMAO and PAGln caused by colonization with the TMA and phenylacetic acid (PAA)-producing species C. sporogenes.
All the impacted metabolites were assessed, along with the metabolic pathways involved in the metabolism of aminoacyl-tRNA biosynthesis, phenylalanine metabolism, tyrosine and tryptophan biosynthesis, arginine biosynthesis, phenylalanine, valine, leucine, and isoleucine biosynthesis, and pantothenate and CoA biosynthesis. Aminoacyl-tRNA biosynthesis enzymes play a role in several metabolic and signaling pathways in addition to their traditional activity. 38 The production of other amino acids, and the structure and function of proteins and enzymes, all depend on the crucial amino acid phenylalanine. The synthesis of proteins requires the aromatic amino acids (AAAs), L-tryptophan, L-phenylalanine, and L-tyrosine. 39 Leucine and isoleucine, the other BCAAs, are produced and degraded by gut microbiota, and the host's circulating levels are linked to type 2 diabetes. 40
Our results suggest that CBD treatment reversed the increase in TMAO and PAGln that was induced by C. sporogenes colonization. This treatment enriched the abundance of beneficial bacteria, such as Lachnospiraceae_NK4A136 and Blautia, in the gut and decreased the levels of cardiovascular risk factors such as TMAO and PAGln. CBD might offer beneficial effects for cardiovascular protection.
Conclusions
To assess the effects of CBD on the metabolic phenotype of the host in the C. sporogenes-transplanted mice, we developed a combined 16S rRNA gene sequencing and UPLC/QTOF-MS-based plasma and fecal metabolomics technique. CBD decreased the levels of CK, ALT, and LDL-C and increased HDL-C markedly, which indicated that it might offer cardiovascular benefits. CBD treatment increased the beneficial genera Lachnospiraceae_NK4A136 and Blautia, but it decreased cardiovascular risk factors such as TMAO and PAGln levels, which suggested that it may have beneficial effects.
Data Availability Statement
Data are available upon reasonable request.
Footnotes
Authors' Contributions
Conceptualization: Y.-J.X., Y.L., and M.H. Methodology: M.H. Formal analysis: M.H., J.S., and A.L.. Data curation: M.H. and J.S. Writing—original draft: M.H. Writing—review and editing: Y.-J.X. All authors have read and approved the article.
Author Disclosure Statement
The authors declare no conflicts of interest.
Funding Information
This work was financially supported by the Key Research and Development Program of Shandong Province (No. 2021CXGC010808), the National Key R&D Program of China (2021YFD2100300), and Collaborative Innovation Center of Food Safety and Quality Control in Jiangsu Province, Jiangnan University.