
Abstract
Introduction:
Medicinal cannabis (MC) has potential therapeutic effects in Tourette Syndrome (TS), however there has been limited research in adolescent patients. This pilot study aimed to investigate the feasibility of conducting a randomized placebo-controlled crossover trial of MC in adolescents with TS.
Method:
This was a phase I/II double-blind, cross-over pilot study comparing MC with matched placebo in adolescents aged 12–18 years with TS. The active medication was Δ9-tetrahydrocannabinol (THC) 10 mg/mL and CBD 15 mg/mL in peppermint-flavored medium-chain triglyceride oil. The dose titration schedule was stratified into two participant weight bands: below 50 kg (max THC 10 mg/day) or ≥50 kg (max THC 20 mg/day). Each treatment phase lasted 10 weeks, with a 4-week washout period.
Results:
Ten adolescents were randomized (mean age 14.8 years, 50% male) and seven completed the full study protocol. Two adolescents discontinued due to adverse events (one on MC, one placebo) and one was lost to follow-up. The most common adverse event was dizziness (67%). There were no serious adverse events. Among actively enrolled participants, protocol adherence was excellent: study visits 100%, blood test completions 100%, and online questionnaire completion 97.6%. Medication adherence was acceptable in 63.6%. Parents reported a high degree of study design acceptability. On the Clinical Global Impression-Improvement scale, three participants were rated as much improved on MC compared with one on placebo at 10 weeks.
Discussion:
The findings suggest that the study protocol is feasible and acceptable to patients with TS and their families. A fully powered study is needed to evaluate the efficacy of MC in adolescent TS.
Introduction
Tourette syndrome (TS) is a neurodevelopmental disorder characterized by motor and vocal tics that have persisted for at least 1 year, with onset before 18 years of age. 1 The prevalence is estimated to be between 0.5% and 1%.2,3 Onset is usually in childhood and the condition typically runs a waxing and waning course and tends to improve with age. 2 Patient with severe tics and/or comorbidities may experience social difficulties, emotional distress, and impairment in quality of life. 4
Recommended management of TS involves a combination of education, behavioral, and pharmacological interventions.5,6 The main classes of medications prescribed for TS are alpha-2 adrenergic agonists and antipsychotics. 7 However, these medications are not effective in all children and often cause significant adverse effects. Hence, alternative treatment options are needed for children with TS who do not respond to or cannot tolerate these medications.
The potential for medicinal cannabis (MC) to treat a range of childhood psychiatric conditions and neurodevelopmental disorders is becoming increasingly understood 8 and there is interest from parents in MC as a treatment. 9 However, there is a lack of evidence to support its use in most conditions, including TS. 10
The two primary cannabinoids are Δ9-tetrahydrocannabinol (THC) and cannabidiol (CBD). Mechanistically, cannabinoids appear to modulate neurotransmission and calcium homeostasis, resulting in potential anti-inflammatory, antioxidant, and neuroprotective effects.11,12 Alterations in the endocannabinoid system have been documented in TS 13 and an association between a gene variant related to the CB1 receptor and risk of TS has been identified. 14 There is a high density of CB1 receptors in brain areas implicated in TS such as the basal ganglia, and these may play a role in motor inhibition. 15 Therefore, MC is a biologically plausible novel pharmacological therapy for TS. 2 Furthermore, there is emerging evidence that MC (particularly CBD) has anxiolytic effects, 16 which may provide indirect therapeutic benefits in TS, as anxiety is commonly a prominent symptom and driver of tics.
To date, the limited research investigating the use of MC in treating TS has largely involved adult participants. Case reports and observational or retrospective studies have suggested MC treatment may reduce tics, urges, compulsive behaviors, and ADHD symptoms.17,18 To date, the potential benefit in TS has been observed with THC, though it has been suggested that treatment may be optimized with the inclusion of CBD rather than THC alone. 19 Improvements in tic severity were reported in three small placebo-controlled trials using THC in dose of 2.5–10 mg/day.20–22 In contrast, a randomized study of single doses of three vaporized MC products and placebo found little effect in video-observed tics in nine adults with TS. 23 Adverse effects reported in these studies included headache, nausea and vomiting, dizziness, hot flushes, ataxia, poor concentration, slowed mentation, and fatigue.
Research on the use of MC to treat TS in children and adolescents is restricted to a few case reports using various products and widely varying doses.24–26 There are currently no registered clinical trials of MC in children or adolescents with TS and there is very limited evidence to inform the use of MC in pediatric TS.
The primary objective of this pilot study was to evaluate the feasibility and acceptability of conducting a placebo-controlled crossover trial of MC in adolescents with TS including the recruitment strategy, tolerability of the study medication, study duration, study procedures, and outcome measures. Secondary objectives were to assess the safety of MC in adolescents with TS and to provide preliminary data on the efficacy of MC compared with placebo on clinical outcomes (tic reduction, clinical global impression) to inform a full-scale randomized controlled trial.
Methods
Trial design
This was a single site, phase I/II double-blind, placebo-controlled, and cross-over pilot study of 10 participants comparing MC with placebo in adolescents aged 12–18 years with TS. Eligible participants were randomized 1:1 to receive either MC in treatment period 1 and matched placebo in treatment period 2 or vice versa.
The active study medication was whole-plant-derived MC, formulated as a solution in medium-chain triglyceride (MCT) oil with a THC concentration of 10 mg/mL and CBD 15 mg/mL. A peppermint flavoring agent was added to disguise any cannabis taste or smell. The placebo was peppermint-flavored MCT oil, indistinguishable from the active medication in smell and taste. The study drug and placebo were provided by Cann Group Limited, Australia. This study received approval from the Human Research Ethics Committee of The Royal Children’s Hospital, Melbourne, Australia.
Participants
This study aimed to enroll a pragmatic sample of adolescents with TS to maximize the generalizability of findings. Therefore, all comorbid disorders other than those listed below were accepted.
Inclusion criteria
Adolescents were eligible if they were aged 12–18 years and had:
DSM-5 diagnosis of TS as assessed by the study clinician. A score of 20 or higher on the Total Tic Severity section of the clinician-administered Yale Global Tic Severity Scale (YGTSS).
27
No changes in either medication or other interventions in the 4 weeks prior to randomization, and intention to remain on the same medications and doses for the duration of the study. Agreed not to drive for the duration of the study.
Exclusion criteria
Non-English-speaking parents.
Participant history of psychosis, schizophrenia, bipolar disorder, or major depressive disorder, or a family history of psychosis.
Taking medications that may interact with MC (e.g., clobazam).
Abnormal liver function tests, defined as alanine transaminase more than twice upper limit of normal.
Use of illicit drugs or MC in the 4 weeks prior to screening.
Pregnant or intending to become pregnant during the study or breastfeeding.
History of clinically significant suicidal thoughts in the prior 12 months.
Study procedures
Recruitment
Participants were recruited from out-patient clinics at the Royal Children’s Hospital, Melbourne, as well as private pediatric practices. The study was advertised to clinicians in relevant departments and private clinics with a request to consider whether they had potentially eligible patients. In addition, the study was advertised via social media from a TS support organization.
Pediatricians and neurologists sent letters to potentially eligible families briefly outlining the study and inviting interested parents to contact the study coordinator for further information. Potential participants then attended a screening visit to determine eligibility. Written informed consent was obtained from parents at the screening assessment.
Randomization and blinding
A randomization schedule was prepared by an independent statistician and provided to the hospital trials pharmacist. Participants were randomized in a 1:1 fashion to THC:CBD 10:15 oil in treatment period 1 and placebo in treatment period 2 or placebo in treatment period 1 followed by THC:CBD 10:15 oil in treatment period 2. Treatment allocation was conducted by the pharmacy who dispensed the study medication according to the randomization schedule. All members of the study team, participants, and their families remained blinded to treatment allocation until the database was locked for analysis.
Dose justification
Dosage of MC was based on the THC component. Although a safe dose for medically prescribed THC is yet to be established, a maximum dose of 30 mg/day has been recommended to minimize the risk of adverse effects. 28 Furthermore, there is some evidence that CBD may provide protection against the psychiatric adverse effects of THC and so it is recommended to use CBD in conjunction with THC. 28 Therefore the available evidence indicates the dose and product chosen should be adequate to identify a clinical benefit, yet carry a low risk of serious adverse events.
Titration and dose optimization
The titration schedule and dose optimization were stratified into two bands depending on the weight of the participant at enrollment: a lower dosage schedule for participants weighing <50 kg and a higher dosage schedule for participants weighing ≥50 kg. Both schedules commenced with 1 mg/day THC, with a protocolized initial up-titration period of 21 days to 5 mg/day for the lower weight schedule, and 10 mg for the higher weight schedule. Once on this dose for 7 days (i.e., Day 29), participants underwent a dose assessment using the Parent Tic Questionnaire (PTQ), 29 to determine whether they had responded to this dose. Participants were considered a responder if they had a 55% reduction from baseline on the PTQ and a Clinical Global Impression-Improvement (CGI-I) 30 score of a 1 or 2, with tolerable side effects. A 55% reduction on the PTQ has been found to be optimal for defining positive treatment response. 31 Responders at the Day 29 assessment remained on that dose for the duration of the treatment period. Participants who had not responded continued up-titrating until they reached 10 mg (lower weight schedule) or 20 mg (higher weight schedule). Medication in both periods was administered once daily unless the participant was unable to tolerate the volume, in which case it was divided into 2 doses/day. Efficacy outcomes were assessed on Day 71 +/− 4 days.
Tapering and washout (days 71–98)
Following post-treatment assessment, participants completed a 7-day three-step down-titration process, followed by a 3-week washout period. A long washout period is recommended for crossover trials of MC due to its accumulation in adipose tissue with slow release. 32
Study schedule
The schedule of study activities is outlined in Supplementary Table S1. There were four study site visits: screening, Day 71, Day 99, and Day 169. At each point of contact, parents were asked about any changes in participants’ concomitant medications. Parents were asked to record each administration of study medication, including administration time, dosage, and any noteworthy comments such as incomplete administration of medication or possible adverse events in a diary. Parents returned medication bottles, empty or otherwise, for pharmacy staff to record the returned volume to measure compliance. Compliance between 80% and 120% was considered acceptable. Study periods in which bottle spillages were reported were excluded from compliance measurement.
Measures
The study measures are described in Supplementary Table S2.
Feasibility of the study design was evaluated by ease of recruitment (adherence to study timeline and rate of recruitment) and percentage completion of study elements. At the conclusion of their involvement in the study, parents completed a questionnaire created by the investigators to assess the acceptability and burden of study procedures, including the recruitment approach, number of study visits, questionnaire completion, completion of blood tests, and medication tolerability. Families were asked in which period they believed the participant was taking the active drug.
Adverse events were recorded at study visits and safety assessment phone calls and rated for severity and relatedness to the study drug. Safety outcomes were also collected using a modified version of the Liverpool Adverse Event Profile (LAEP), 33 completed by the parent. The LAEP was designed to capture known side effects of antiepileptic medication. The modified version includes additional items to ascertain other known side effects of MC. A two-point increase in symptoms on this measure was considered clinically meaningful. Liver function tests were repeated at the end of each treatment period.
Data collection and analysis
All data were entered into a study-specific online database (REDCap, Vanderbilt University, TN). 34 Feasibility outcomes are presented for all participants’ expected study elements prior to being recorded as completed, withdrawn or lost to follow up. Safety and efficacy outcomes are presented by treatment condition (active or placebo) as summary data. To assess for a signal of efficacy, data from the two main clinical outcome measures, YGTSS Total Tic score and the CGI-I, were analyzed and reported. Change scores on the YGTSS Total Tic Score were calculated by comparing baseline scores with scores at the end of the treatment period for each condition, presented as a mean change and its standard deviation (SD). Individual change scores were also reported, interpreted relative to the SD (8.62) of the Total Tic Score in children and adolescents with TS. 31 On the CGI-I, we examined the proportion of participants rated as much improved or very much improved by treatment condition. This pilot study was not powered to determine efficacy and so results of these clinical outcome measures are presented descriptively only.
Results
From April 2022 to August 2023, 17 adolescents were prescreened for enrollment. Seven of these were excluded due to being ineligible. Reasons for exclusion included tic severity below threshold (n = 2), comorbid significant mental health concerns (n = 4) and a close family history of psychosis (n = 1). The remaining 10 were invited to attend a clinic screening appointment. Of those, all 10 met eligibility criteria and consented to participate and so were randomized to treatment. Five were randomized to receive MC in the first treatment period and five were randomized to receive MC in the second treatment period. All received treatment in the intended order. Demographic and clinical characteristics of study participants are described in Table 1.
Demographic and Clinical Characteristics of Study Participants at Screening (n = 10)
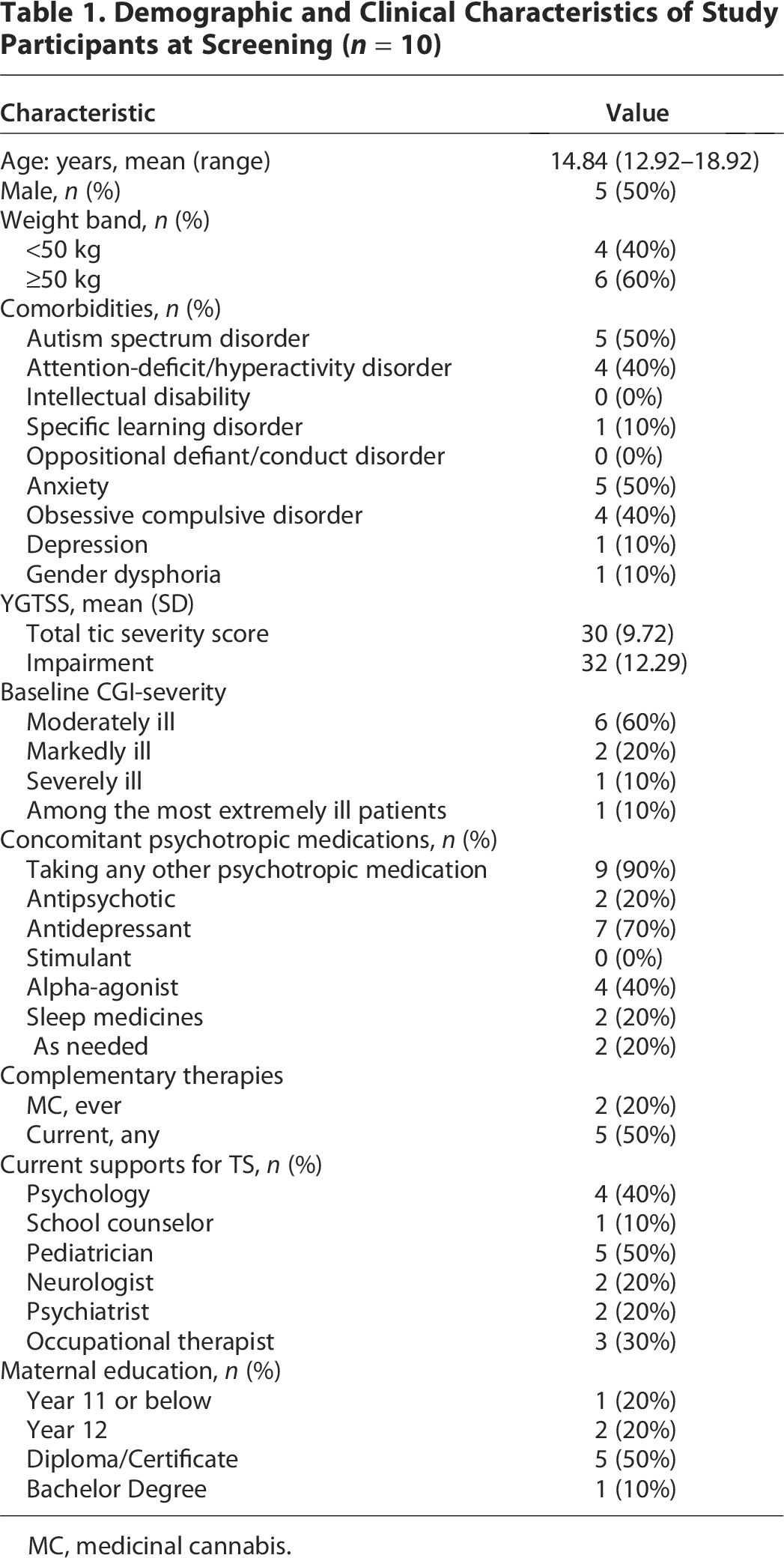
MC, medicinal cannabis.
Seven out of 10 participants completed the study protocol as intended. One participant discontinued study drug during treatment period 2 (MC) due to an adverse event, but remained in follow-up and completed the outcome measures at the intended time points. One participant withdrew from the study during treatment period 1 (placebo) due to an adverse event and declined to attempt treatment period 2. One participant was lost to follow up during treatment period 1 and hence no post-treatment outcome data are available. Complete datasets were available for clinician-rated outcome measures for eight participants, and for parent-rated outcome measures for seven participants.
At Day 29 dose assessment, only one participant reported clinically significant improvement at the lower dose—this participant was in the higher weight dosing schedule and taking MC during that treatment period. All other participants continued up-titration after Day 29.
Feasibility
During the 16-month recruitment phase, participants were recruited at a rate of one participant every 7 weeks, which was in line with the study targets. Table 2 outlines protocol adherence for key elements of the study design. During the trial, some participants reported that the active drug and placebo had a slight difference in color, however, only some participants correctly guessed which was the active drug. For two participants, study drug compliance calculations were deemed unreliable due to known spillage and therefore not reported.
Protocol Adherence
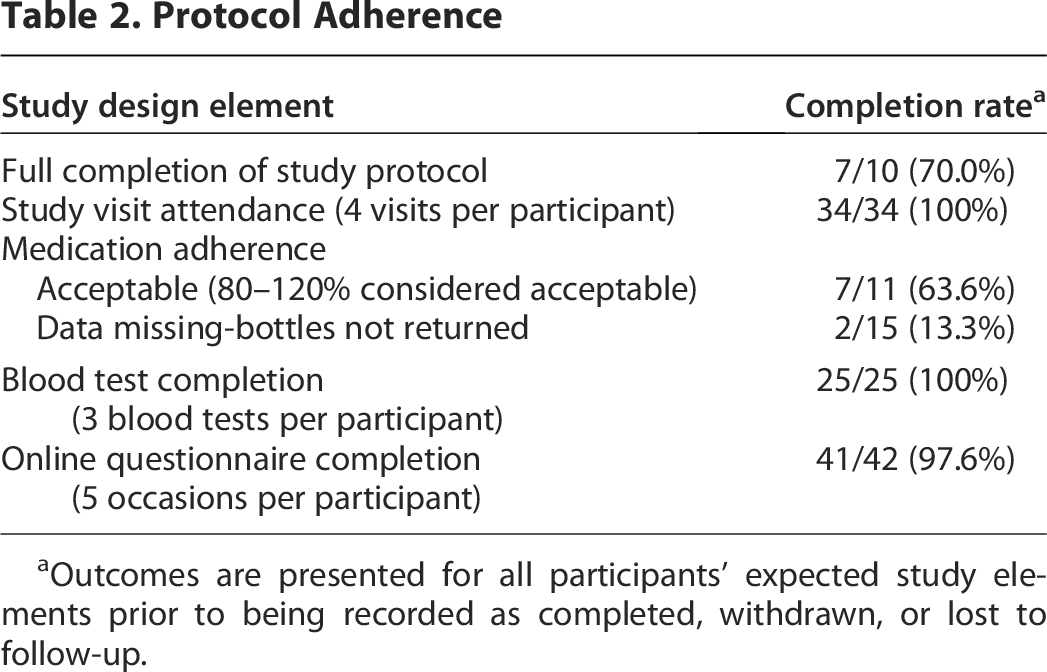
Outcomes are presented for all participants’ expected study elements prior to being recorded as completed, withdrawn, or lost to follow-up.
Acceptability
Eight parents completed the poststudy evaluation questionnaire. Acceptability ratings were generally high (Table 3).
Pilot Acceptability Evaluation—Parent Responses
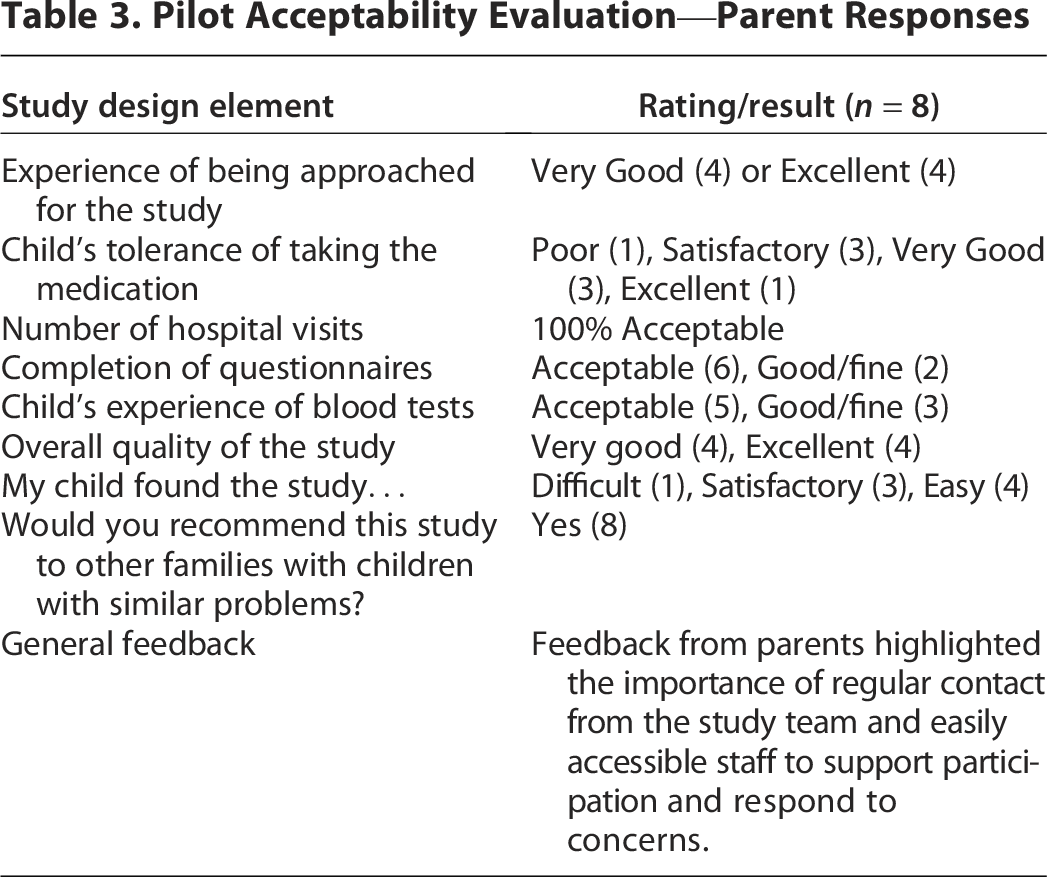
Safety
The medication was generally well-tolerated. The most frequently reported adverse event was dizziness (Table 4). Two participants required the dose to be split into twice daily dosing due to adverse events both in the MC condition (one with panic symptoms and dizziness, one with fatigue). In one case, symptoms resolved after dose splitting, however, the other required a dose reduction (0.5 mL rather than 1 mL). There were no severe or serious adverse events considered to be related to the study drug and no clinically significant abnormal laboratory test results while taking MC. Two patients discontinued treatment due to presumed adverse events, one while taking MC and one while taking placebo—in both cases fatigue. All adverse events considered potentially related to the study drug resolved, either during treatment or after ceasing treatment. Three participants had a change in body weight of >5% from baseline to the end of the treatment period—all taking placebo at the time. The proportions of participants who experienced a two-point increase from baseline to end of each treatment period on the parent-reported LAEP are presented by symptom in Supplementary Table S3.
Adverse Events

Denominator in each treatment condition was 9, as one participant did not commence the active drug period and one did not commence the placebo period.
Efficacy outcomes
Mean reduction in Total Tic Score (YGTSS) was higher in the active drug period (−3.9; SD 8.8) compared with placebo (0.0; SD 5.9). Three participants had reductions in Total Tic Scores in the order of one SD or more (7, 9, and 20 points) on active drug compared with one on placebo (12 points). On the CGI-I 3/8, participants (38%) were rated as much improved or very much improved in the MC period compared with 1/8 participants (13%) in the placebo period (Table 5).
Efficacy Outcomes

PTQ, Parent Tic Questionnaire; SD, standard deviation.
Discussion
This pilot study evaluated the feasibility and acceptability of conducting a randomized, double blind, placebo-controlled study of oral MC in adolescents with TS. Results indicate that the study design is feasible in this patient population. Parents reported high levels of acceptability, with all indicating they would recommend the study to other families with children with similar clinical problems. The most common adverse event during MC treatment was dizziness, reported by two-thirds of participants.
The efficacy data provide preliminary evidence in favor of MC as a potential treatment for adolescent TS, with a larger mean change in YGTSS Total Tic score on MC compared with placebo, and more participants rated as much improved on the CGI-I when on MC. Although the numbers were very small, these data are consistent with adult studies of MC in TS20,21 and support the need for a fully powered randomized controlled trial to test the efficacy of MC in the pediatric TS population.
This study had some limitations. First, the placebo was not well-matched for appearance. This was, however, unlikely to have influenced the findings as the participants were not able to tell which was the active drug based on appearance. Second, the proportion of participants with comorbid autism spectrum disorder (50%) was higher than is typical in TS samples (21–23%), 35 and this may have affected the findings. Third, as this was a small pilot study (and three participants did not complete the full protocol), the efficacy signal and safety data need to be interpreted with caution. However, this study provides valuable insights into feasibility and acceptability, providing proof of principle for potential efficacy. Finally, we used a product with a 2:3 ratio of THC:CBD, with a ceiling THC dose, and so cannot comment on the tolerance or effect of alternate MC products or higher doses.
In summary, the findings from this pilot study suggest that our protocol is feasible and acceptable to adolescents with TS and their families. Further research is recommended to expand on these initial findings in a larger cohort, powered to evaluate efficacy and investigate whether subgroup characteristics are associated with variation in treatment response, e.g., baseline severity, comorbidity profile, and age band. In addition, given the potential risks of exposure to THC, dose finding should be an area of future research to try to identify the minimal effective dose. If proven effective and safe, MC could provide a novel treatment option for adolescents with TS. It should be noted that if adolescents are exposed to cannabis for longer periods of treatment then follow-up evaluation for potential negative cognitive and psychiatric effects would be important.
Authors’ Contributions
All authors made substantial contributions to the design of this study and the writing of the protocol, made substantial contributions to the acquisition, analysis, or interpretation of data for the work; made substantial contributions to drafting the work and revising it critically for intellectual content; approved the final version submitted; agree to be accountable for the accuracy or integrity of the work.
Footnotes
Author Disclosure Statement
D.E. reports grants and nonfinancial research support from Cannatrek Medical Pty Ltd and nonfinancial research support from THC Pharma Pty Ltd and Tilray. P.I.L. receives funding support from Patient-Centered Outcomes Research Institute, USA. K.J.L. was supported by an investigator grant from the Australian National Health and Medical Research Council (ID 2017498). All other authors have no interests to disclose.
Funding Information
This work was supported by a small grant from Cann Group, who also supplied the investigational product in-kind. Cann Group had no role in the conception or design of the study; data collection, management, analysis, or interpretation; preparation, review, or approval of the article; nor in the decision to submit the article for publication. This research was also supported by the Victorian Government’s Operational Infrastructure Support Program.
Declarations
The authors confirm that the Principal Investigator for this article is D.E. and that he had direct clinical responsibility for patients. This study received approval from the Human Research Ethics Committee of The Royal Children’s Hospital, Melbourne, Australia (19th February 2021, HREC reference number 69238). This study was prospectively registered with ClinicalTrials.gov on 11 Jan 2022 (ID NCT05184478).
Data Availability Statement
The data from this study are available from the corresponding author upon reasonable request. A data access agreement must be signed between relevant parties. Data will only be shared with a recognized research organization that has approved the proposed analysis plan.