
Abstract
Introduction:
In epidemiological studies, people who use cannabis have a lower prevalence of obesity. Furthermore, the endocannabinoid system is recognized as a potential target for obesity treatment and partial agonism of the cannabinoid type-1 (CB1) receptor may reduce body weight. We thus hypothesized that 12 weeks of pharmacotherapy with the partial CB1 receptor agonist nabilone would reduce body weight, relative to placebo, in adults with obesity.
Methods:
We conducted a randomized, double-blind, placebo-controlled pilot clinical trial that investigated the feasibility, tolerability, and efficacy of 12 weeks of treatment with nabilone compared with placebo in adults with obesity. Otherwise healthy adults aged 25–45 years with obesity were randomized in a 1:1:1 ratio to one of three parallel treatment arms: high-dose nabilone (6 mg/day), low-dose nabilone (2 mg/day), or placebo. Safety and feasibility outcomes included adverse events (AEs), number of dropouts, and medication adherence per treatment arm. Efficacy outcomes included body weight, body mass index (BMI), and waist circumference. Secondary outcomes included gut microbiome changes, blood biomarkers (e.g., glucose and insulin levels), and mood.
Results:
Overall, 18 participants were randomized and 15 participants received at least one dose of drug (4 high-dose arm, 5 low-dose arm, 6 placebo). The trial was terminated early due to poor tolerability of the medication (e.g., all four participants allocated to high-dose nabilone withdrew due to AEs). Only eight participants completed per protocol (four in the low-dose arm and four in the placebo arm). Using data from completers only (n = 8), we saw a significant treatment effect on body weight (p < 0.001) and BMI (p < 0.001) that appeared to be driven by greater decreases in the low-dose arm (n = 4) relative to placebo (n = 4). Based on the Bray–Curtis dissimilarity, the low-dose arm showed a greater change in the overall fecal microbiome composition compared with the placebo arm (p < 0.05).
Discussion:
This pilot trial found poor tolerability of nabilone pharmacotherapy (especially at 6 mg/day) for adults with obesity who had not used any cannabinoid drugs for 6 months prior to enrolment. Preliminary results suggest a possible impact of nabilone on the gut microbiome.
Introduction
Obesity is defined as a body mass index (BMI) greater than 30 kg/m2 (though note this definition may change, see Ref. 1 ) and is a serious health concern due to its association with decreased quality of life 2 and increased risk of medical conditions such as coronary heart disease and type 2 diabetes. 3 Between 1990 and 2022, age-standardized rates of obesity increased worldwide in nearly all of the 200 countries with available estimates, with an overall doubling of the prevalence of obesity in adults. 4
The endocannabinoid system (ECS) has been recognized as a potential target for obesity treatment since the early 2000s, given its role in appetite regulation and energy balance both centrally and peripherally.5,6 Evidence that excessive activation of the ECS contributes to the pathogenesis of obesity led to clinical trials testing the anti-obesity potential of ECS antagonism,5,7 with a focus on rimonabant, an inverse agonist of the cannabinoid type-1 (CB1) receptor. While rimonabant significantly reduced body weight and improved secondary measures such as dyslipidemia and cardio-metabolic risk factors in adults with obesity,8,9 it was ultimately withdrawn from the market due to serious psychiatric adverse effects, including increased risk of suicidal ideation and depression.10–12 The therapeutic promise of rimonabant fueled ongoing efforts to identify other CB1 receptor ligands that might have potential to reduce body weight and improve health outcomes in obesity.7,13
Despite the association between ECS activation and obesity, there is mounting evidence that chronic exposure to delta-9-tetrahydrocannabinol (THC), a primary psychoactive component of cannabis and a partial agonist at the CB1 receptor, is associated with reduced body weight and risk of obesity. 7 Using data from the National Epidemiological Survey on Alcohol and Related Conditions and the National Comorbidity Survey-Replication study, our group found that the rate of obesity was significantly lower in individuals reporting frequent (more than three times per week) cannabis use compared with individuals who did not use cannabis in the last 12 months. 14 These findings have been replicated in adolescents 15 and young adults 16 and in prospective, longitudinal research. 17 Further, we showed that chronic THC treatment reduced weight gain, fat mass gain, and energy intake in diet-induced obese mice, possibly by preventing changes in gut microbiota. 18
The goal of this pilot randomized controlled trial (RCT) was to determine if a partial CB1 receptor agonist has the potential to reduce body weight and improve related health outcomes in adults with obesity. We chose nabilone, a synthetic analogue of THC, because it is a partial agonist at the CB1 receptor with slightly greater efficacy than THC. 19 Our primary aim was to determine the feasibility (number of completers and medication adherence) and tolerability (number of adverse events [AEs]) of 12 weeks of daily treatment with nabilone in adults with obesity who had not used any cannabinoid drugs for 6 months prior to enrolment. Our secondary aims focused on exploring preliminary efficacy (e.g., reduction of body weight) and potential underlying mechanisms (e.g., changes in metabolic biomarkers and gut microbiota) by comparing the nabilone-treated groups to the placebo-treated group. Since nabilone has been well tolerated in trials with other patient populations (e.g., in cannabis dependence 20 and post-traumatic stress disorder 21 ), we hypothesized that nabilone would be well tolerated, with few serious AEs, and would promote weight loss, relative to placebo.
Methods
We conducted a randomized, double-blind, placebo-controlled pilot RCT that investigated the feasibility, tolerability, and preliminary efficacy of 12 weeks of treatment with nabilone compared with placebo in adults with obesity. Participants were randomized in a 1:1:1 ratio to one of three parallel treatment arms: high-dose nabilone (6 mg/day), low-dose nabilone (2 mg/day), or placebo. The trial took place at a single site, the Center for Addiction and Mental Health (CAMH) in Toronto, Ontario, Canada and recruitment occurred between February 2022 and August 2023 (see Supplementary Data for more recruitment details). The trial was approved by the CAMH Research Ethics Board (REB # 084/2018) and was registered on ClinicalTrials.gov (ID NCT04801641).
Participants
To be enrolled in the trial, female or male adults aged 25–45 years had to meet the WHO definition of obesity (BMI >30 kg/m2), have liver and kidney function within normal limits at screening, and agree to use adequate methods of contraception (as relevant). Additionally, the following criteria excluded participation: unstable gastrointestinal, respiratory, endocrinological, cardiovascular or cerebrovascular disease; unstable major psychiatric disorder(s); current substance use disorder (DSM-5) (excluding tobacco and caffeine); history of, or current neurological illnesses; current use or use during the previous month of antipsychotic medications; learning disability, amnesia or other conditions that impede memory and attention; visual impairments; personal or family history of schizophrenia, or psychosis (or psychosis-related) disorders; antibiotic use in the last 4 weeks; previous bariatric surgery; current use or use in the past month of other weight-loss pharmaceuticals; cannabis use in last 6 months; known sensitivity to cannabis or other cannabinoid agents; and pregnancy or lactation.
Intervention
The CAMH research pharmacy purchased and over-encapsulated generic nabilone (0.5 mg pms-nabilone capsules) and prepared matching placebo capsules. The nabilone dose in the two active medication arms was titrated up to 2 mg/day (1 mg twice a day, in the morning and at bedtime) over a 1-week period: two nabilone capsules at bedtime for 3 days then addition of two nabilone capsules in the morning for the following 4 days. Thereafter, low-dose participants were maintained on the 2 mg/day regimen and high-dose participants were escalated to 6 mg/day (3 mg, twice a day) with the addition of four nabilone capsules at bedtime for 3 days then addition of four nabilone capsules in the morning for the following 4 days. All participants took six capsules in the morning and six at bedtime (a mix of active and/or placebo, as needed, for proper blinding). The CAMH pharmacy dispensed 1 week’s supply of medication at a time. At the beginning of the trial, all participants received standardized diet and exercises strategies to promote weight loss and complement the pharmacotherapy.
Once participants were enrolled and scheduled for their first weekly visit, the CAMH pharmacy randomized participants into one of the three medication arms. The pharmacy used two randomization tables with 30 codes in each, one table for females and one for males, both with two blocks of 15. In each block, the assignments were equally distributed between medication arms (1:1:1). Participants and the research team (excluding the pharmacy) were blinded to medication arm. During the final study visit, we administered a questionnaire asking participants whether they felt they had received active or placebo medication.
Study procedures
Following telephone prescreening, participants were invited onsite for a screening visit, which included a medical exam, blood work, and the Structured Clinical Interview for DSM-5 (SCID-5-RV) to screen for any potential mental health or substance use disorders. Baseline data were collected at the screening visit. Eligible participants were enrolled and attended a brief post-enrollment visit and then 13 weekly visits. At each weekly visit, a 1 week’s supply of medication was dispensed and the following measures were collected: vital signs, height/weight and waist measurement, urine toxicology screen, urine pregnancy screen (females), Hamilton Depression Rating Scale (HAM-D), 22 Beck Depression Inventory (BDI), 23 and Eating Attitudes Test (EAT-26). 24 Blood samples were drawn at Weeks 1, 5, 9, and 12. A fecal sample kit was given to participants at baseline and at Week 11, and then collected at Week 1 and Week 12 visits, respectively.
Outcomes
Our preregistered co-primary outcomes were: (1) number of serious AEs (SAEs; measure of safety/tolerability) and (2) number of dropouts (measure of feasibility), per treatment arm and over the course of the 12 weeks of treatment. AEs were captured using the Systematic Assessment for Treatment Emergent Effects (SAFTEE), a structured tool that ascertains the presence of common somatic, behavioral, and affective symptoms. 25 Medication adherence was based on pill counts and was operationalized as the total number of capsules returned at each weekly visit (since participants took six capsules twice a day, six returned capsules corresponds to one missed dose, and 12 returned capsules corresponds to 1 day of missed medication). Preregistered secondary outcomes included body weight (kg), gut microbiota (based on fecal samples; see Supplementary Data for details), and blood levels of glucose (random glucose, mmol/L), insulin (pmol/L), triglycerides (mmol/L), total cholesterol (mmol/L), high-density lipoprotein (HDL) cholesterol (mmol/L), and low-density lipoprotein (LDL) cholesterol (mmol/L). We had planned to include two additional imaging measures (abdominal fat using structured magnetic resonance imaging [MRI] and neural reactivity to food stimuli using functional MRI), but we did not have any participants complete both the baseline and end-of-study imaging procedures, so there are no data to report (the imaging component of the study was optional, and few participants consented to this component). Similarly, we collected blood samples for additional biomarker measures (leptin, ghrelin, and PYY), but unfortunately the samples were lost in transportation.
Sample size
Results from previous studies were combined for the power calculation: preclinical data showing the efficacy of both THC and rimonabant in reducing food intake and preventing diet-induced obesity18,26 and data from a controlled trial of rimonabant for weight reduction in humans. 27 The significance level was set at 0.05. We determined that to detect an effect equivalent to a 3% drop in body weight in the high-dose nabilone arm compared with a 1.8% drop in the placebo arm with 45% power, we would require 20 completers in the nabilone arm. Thus, our proposed sample size was 60 completers (20 per treatment arm). Ultimately, we did not reach our target sample size, as the trial was terminated early due to an unanticipated number of dropouts related to AEs.
Statistical analysis
All data analysis was conducted in SPSS v27. Baseline data are presented descriptively using means and standard deviations (SDs). Baseline data for all randomized participants are presented in Supplementary Table S1 in Supplementary Data and in Table 1 for completers only. Independent samples t-tests were used to determine if there were significant differences between the two groups of completers. In order to provide estimates of nabilone efficacy, we analyzed relevant outcome measures over the 12 weeks of treatment in completers only (n = 4 low-dose group and n = 4 placebo) using split-plot analysis of variance, where time was a within-subjects factor and group was a between-subjects factor. Where any time by group interactions were significant, we present the mean and 95% confidence intervals (CIs) for the baseline value and the Week 13 value for each group.
Participant Demographics and Baseline Characteristics, by Medication Arm
No continuous baseline measures were statistically different between groups based on independent-sample t-tests.
SD, standard deviation; F, female; M, male; BDI, Beck Depression Inventory; HAM-D, Hamilton Depression Rating Scale; EAT-26, Eating Attitudes Test; BSQ, Berlin Sleep Questionnaire.
Results
Participant characteristics and retention
Thirty-seven participants were screened onsite; 13 were excluded (8 ineligible; 5 only partially completed screening and did not return) and 24 were enrolled. Six enrolled participants were lost before randomization, so 18 participants were randomized: five to the high-dose arm, six to the low-dose arm, and seven to placebo. One participant from each arm did not receive drug due to scheduling issues (i.e., missed the first weekly visit and did not return). All four remaining participants in the high-dose condition withdrew from the study due to AEs, while one participant withdrew from the low-dose condition due to AEs, and two from the placebo arm for the same reason. Thus, of the 18 randomized participants, only 8 (3 female participant, 5 male participants) completed the trial: 4 in the low-dose arm and 4 in the placebo arm (see Figure 1).
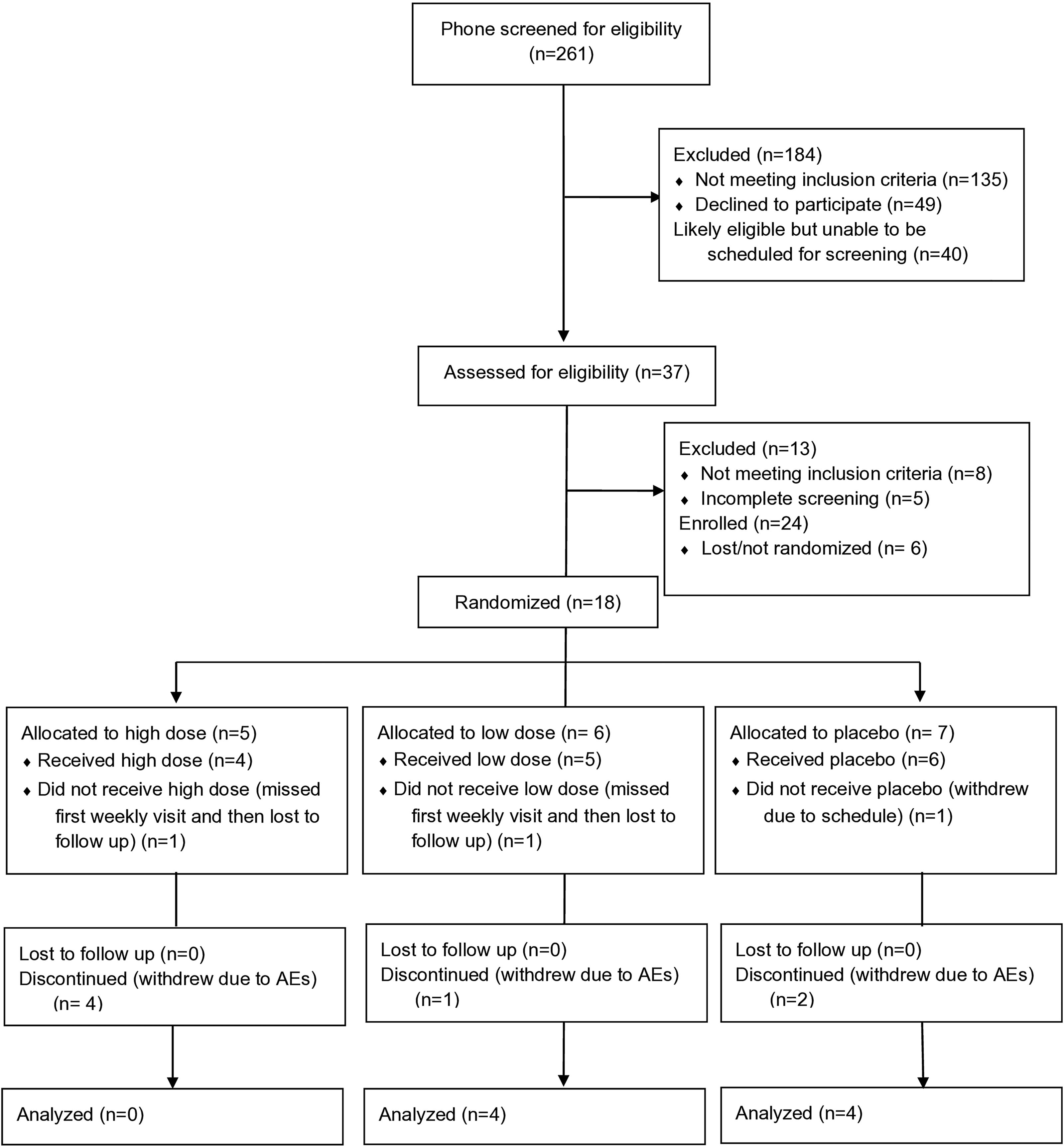
Participant flowchart following the CONSORT 2010 template. CONSORT, Consolidated Standards of Reporting Trials.
Table 1 presents baseline characteristicsfor completers (see Supplementary Table S1 in Supplementary Data for baseline characteristics of participants that started medication). Overall, completers were mean (SD) 33.1 (7.4) years old, had a baseline BMI of 41.2 (7.3) kg/m2, were 120.3 (18.7) kg, and had a waist circumference of 127.7 (18.0) cm. The mean baseline BDI score was 7.1 (5.4), indicating mild depression, 23 HAM-D was 3.6 (2.3), which is in the “normal” range (no clinical evidence of depression), 22 and EAT-26 was 7.8 (5.4), which is well below the threshold (20) for a suspected eating disorder. 24
Based on pill counts, five completers (three in the low-dose arm, two in the placebo arm) took all capsules as instructed. One completer in the lose-dose arm missed six capsules during Week 3, one completer in the placebo arm missed 12 capsules during Week 8, and one additional participant in the placebo arm missed 54 capsules total (12 during Week 3, 12 during Week 5, 18 during Week 10, and 12 during Week 12). In contrast, in the high-dose arm, all participants withdrew and stopped taking medication within the first 3 weeks of treatment and all missed at least 48 capsules each week before they withdrew, with the exception of one participant who took all 84 capsules during Week 1.
Safety and tolerability
There were no SAEs over the course of the trial. However, all but one participant who was randomized and received medication (14 out of 15) experienced at least one AE, leading to 76 total AEs recorded (note that multiple discrete occurrences of the same AE were counted as separate AEs for each participant). Of these 76 AEs, 26 occurred in the high-dose group, 33 in the low-dose group, and 17 in the placebo group. It is important to note that all four participants who received medication in the high-dose group withdrew within the first 3 weeks of treatment, so there was a disproportionate number of AEs reported in this group. The most commonly reported AEs were dry mouth (11 occurrences), drowsiness (7 occurrences), increased appetite (7 occurrences), decreased appetite (6 occurrences), and dizziness (6 occurrences). All of these AEs were common reasons for withdrawing in the high-dose group, with the addition of intoxication that was reported three times (by two participants) in this group. AEs are tabulated for all randomized participants by medication group in Table 2. We note that the high prevalence of AEs is likely due to the structured SAFTEE tool that we used, in which participants were prompted to inform the team if they had experienced any symptoms from a list of common somatic, behavioral, and affective symptoms at each weekly visit. Structured assessments are known to elicit greater AE endorsement than spontaneous assessments, as participants often misattribute commonly occurring symptoms (e.g., drowsiness, headache) to the intervention (including placebo). 28 Additionally, a recent umbrella review documented that across clinical populations, dropouts in cannabinoid trials due to expected AEs associated with acute cannabinoid effects were not uncommon. 29
Incidence of Adverse Events for All Randomized Participants That Received Medication (n = 15), according to Medication Arm
Mild and moderate severity are grouped together; no adverse events were severe.
AEs, Adverse events.
Efficacy
For our primary efficacy outcome, body weight (Fig. 2a), there was a significant medication group by time interaction, F(13,78) = 6.0, p < 0.001. The low-dose group saw a reduction in body weight over the 12 weeks (mean baseline [95% CI] weight 120.3 [95.6–145.0] kg vs. Week 13 weight 111.5 [87.3–135.6] kg) while the placebo group saw minimal change (a slight increase in body weight) over the 12 weeks (baseline weight 120.3 [95.6–145.0] kg vs. Week 13 weight 124.5 [100.3–148.6] kg). Similarly, for BMI (Fig. 2b), there was a significant medication group by time interaction, F(13,78) = 4.6, p < 0.001. In the low-dose group, there was a reduction in BMI over time (baseline BMI 43.4 [34.2–52.5] kg/m2 to Week 13 BMI 40.2 [31.8–48.6] kg/m2), while the placebo group saw minimal change (baseline BMI 38.9 [29.8–48.0] kg/m2 to Week 13 BMI 39.4 [30.9–47.8] kg/m2). Finally, there was no significant medication group by time interaction for waist circumference (Fig. 2c), F(13,78) = 1.5, p = 0.13.
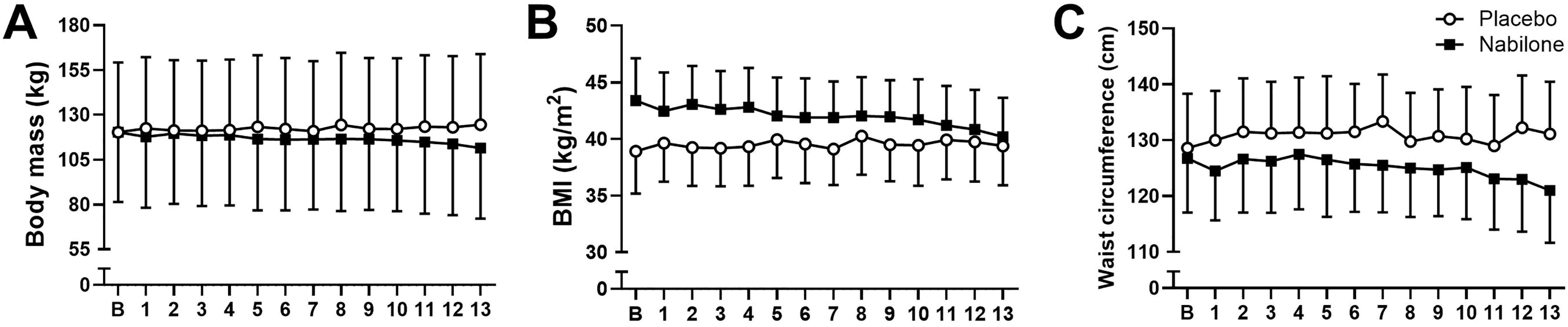
Exploratory efficacy measures plotted for the low-dose nabilone arm (n = 4) and the placebo arm (n = 4) from the baseline visit
Gut microbiota
Beta diversity metrics, including Bray–Curtis dissimilarity and Aitchison distance of the amplicon sequence variants, were visualized using a principal coordinate analysis and redundancy analysis, respectively (Fig. 3A, B). Based on the Bray–Curtis dissimilarity, patients who received a low dose of nabilone showed a greater change in the overall fecal microbiome composition between baseline and Week 11 compared with those in the placebo group T(5) = 3.4, p = 0.02 (placebo, 0.239 [0.205–0.272] vs. low-dose, 0.406 [0.314–0.497]).; however, no statistical difference was seen for Aitchison distance, T(5) = 0.74, p = 0.49 (placebo, 42.2 [38.0–46.3] vs. low-dose, 46.6 [35.6–57.7]) (Fig. 3C–D). The fecal microbiome of patients at baseline showed no significant differences in alpha diversity compared with Week 11 in both the placebo (linear mixed-effects model (lmm), estimate = 0.11, 95% CI [−0.0280, 0.251], p = 0.18) and low-dose nabilone groups (lmm, estimate = 0.012, 95% CI [−0.190, 0.214], p = 0.90) (Fig. 3E).
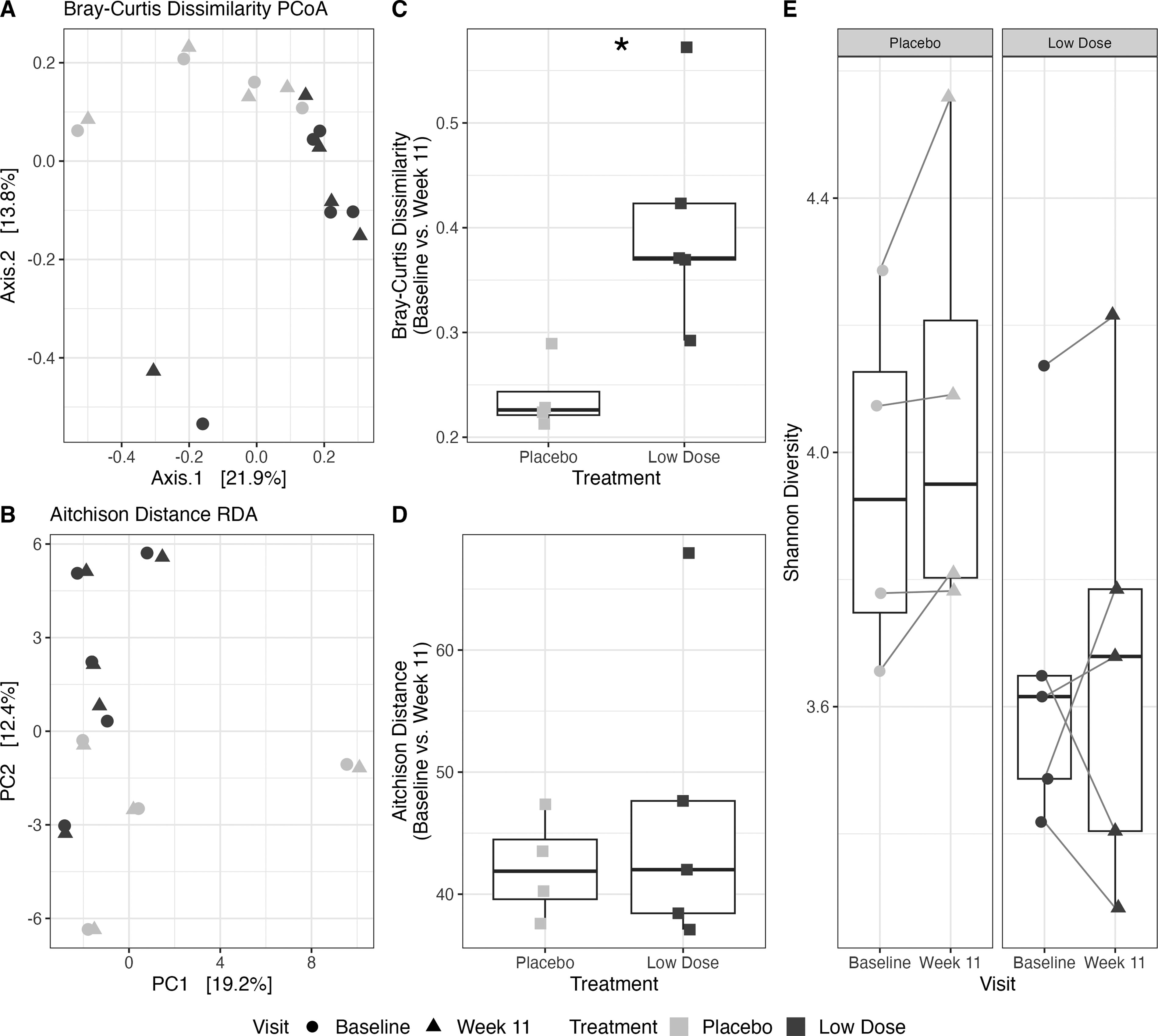
Comparison of the fecal microbiome at baseline and following placebo or low-dose nabilone (Week 11) as a treatment for obesity using 16S rRNA amplicon sequencing. Changes in the diversity of the fecal microbiome were assessed by extracting genomic DNA and performing 16S rRNA sequencing analysis on patient fecal samples from the placebo (n = 4) and low-dose nabilone (n = 5) groups. Beta diversity of the ASVs was visualized using
Other outcomes
There was a significant medication group by time interaction for BDI scores (Fig. 4a), F(13,78) = 1.9, p = 0.048. Mean BDI scores decreased over time in both groups, though the decrease was quantitatively larger in the placebo group (low-dose baseline BDI score, 4.8 [−1.6–11.1] to Week 13, 4.2 [0.3–8.2]; placebo baseline BDI score, 9.5 [3.2–15.8] to Week 13, 1.2 [−2.7–5.2]). There was a similar group by time interaction for HAM-D scores (Fig. 4b), F(13,78) = 1.8, p = 0.048. HAM-D scores actually increased over time in the low-dose group (baseline, 3.5 [0.4–6.6] to Week 13, 6.2 [2.1–10.4]) while scores decreased in the placebo group (baseline, 3.8 [0.7–6.8] to Week 13, 0.5 [−3.7–4.7]). There was no significant interaction or significant main effects of dose or time for EAT-26 scores (Fig. 4c). Similarly, there were no significant condition by time interactions or main effects of condition for any of the laboratory measures (glucose, insulin, triglycerides, total cholesterol, HDL, or LDL; see Fig. 5). However, there was a significant main effect of time for HDL cholesterol, F(3,9) = 8.4, p = 0.006, where HDL decreased over time in both groups.

Secondary safety measures screening for depression and eating disorder risk plotted for the low-dose nabilone arm (n = 4) and the placebo arm (n = 4) from the baseline visit
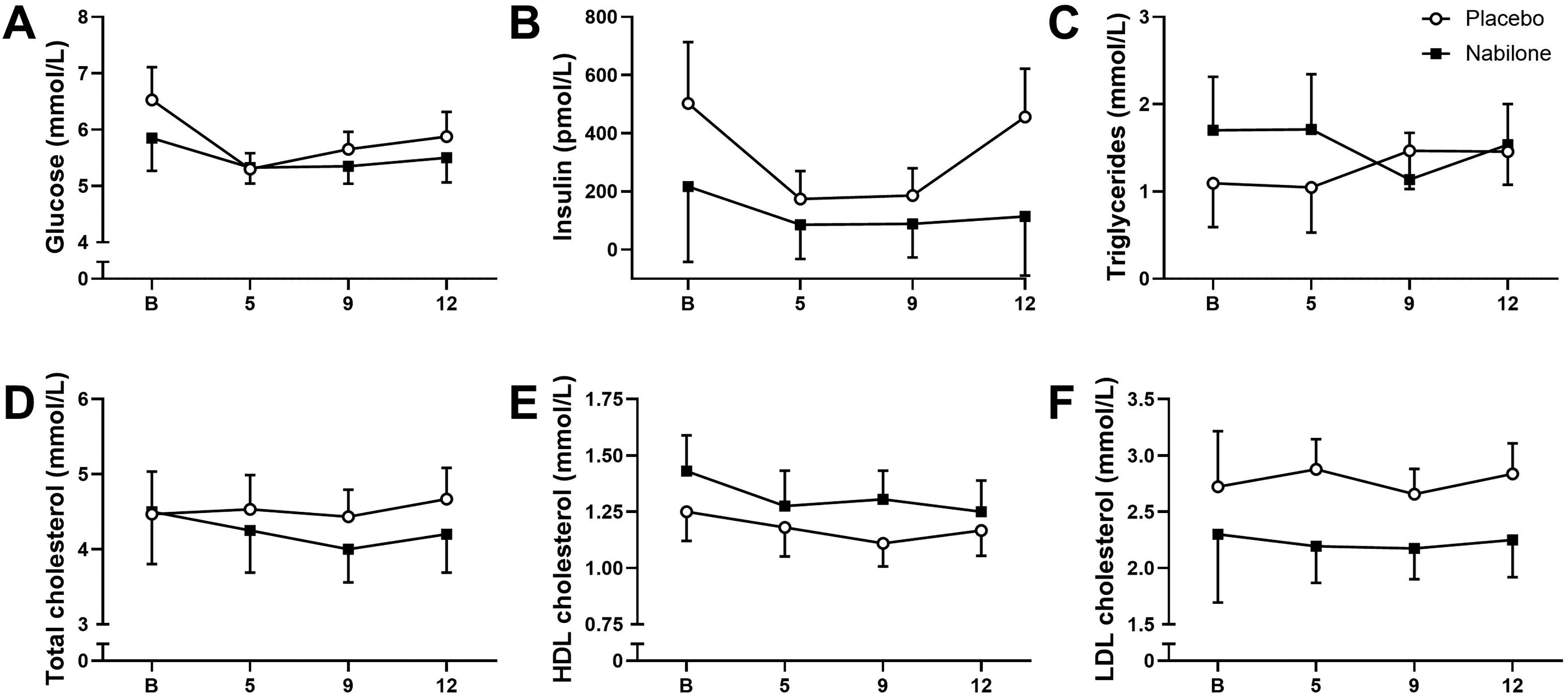
Secondary laboratory measures plotted for the low-dose arm (variable n due to missing data) and the placebo arm (variable n due to missing data) at the following visits: baseline visit
Discussion
The goal of this study was to explore for the first time if a partial agonist of the CB1 receptor (nabilone) could reduce body weight and improve related outcomes in adults with obesity. Our trial suggested poor tolerability of 12 weeks of nabilone pharmacotherapy (especially at the highest dose) in participants who had not been exposed to cannabinoids for the 6 months prior to enrolment, as we terminated the trial early due to the number of AE-related dropouts. Our target sample was 20 in each medication group, yet zero completed in the high-dose group and just four participants completed in both the low-dose and placebo groups. Medication adherence was very poor in the high-dose group (all participants randomized to this group withdrew due to AEs within the first 3 weeks of treatment), though adherence was notably better in the low-dose group. AEs like dry mouth, drowsiness, dizziness, and intoxication (high-dose group only) were reported by participants as reasons for withdrawing early from the trial, suggesting poor tolerability of the medication, especially at the high dose. Two participants in the high-dose group withdrew and stopped taking their medication during the first week of treatment, which is an up-titration week in which the participants are not receiving the full dose of the medication, suggesting that even lower doses were poorly tolerated in some participants. It should be noted that we recruited participants who had not used cannabis in the past 6 months and had minimal lifetime exposure to cannabis, which was intended to mitigate tolerance for this class of medication and ensure that any observed outcomes after trial enrolment were due to the investigational drug. It is possible that a lower dose initiation and more gradual dose increase could lead to better tolerability of nabilone in this population. Further, since participants noted to the staff that they did not like the feeling of being intoxicated, it is possible that nabilone might have better tolerability in a sample of patients with greater previous exposures to cannabis or other cannabinoid drugs. In sum, the results of our pilot RCT did not support our hypothesis of good tolerability of nabilone and do not support nabilone as a clinically useful treatment for obesity in adults, at least at the doses tested and in this selected population.
Despite the early termination of our trial and the evidence of poor tolerability of nabilone (especially at the higher dose), we did see some evidence of preliminary efficacy based on data from completers only. There was evidence of a decrease in body weight and BMI in the low-dose nabilone group over the 12 weeks of treatment (decrease of about 9 kg of body weight and 3 kg/m2 BMI), while there was minimal change in the placebo group. This evidence supports our initial hypothesis that nabilone treatment would lead to a reduction in body weight and is consistent with preclinical literature that has found chronic THC treatment reduces body weight in rodent models and with epidemiological literature finding lower BMI in adults and adolescents who use cannabis. 7 However, due to the very small sample size, those results should be interpreted with caution. There was also some evidence of changes in mood over the course of the trial, particularly an increase in HAM-D scores in the low-dose group. However, mean scores were consistently in the “normal” range (below 7), indicating no clinical evidence of depression, 22 and no participants reported any mood-related AEs during the course of the study.
While our sample size was ultimately too low to comment on potential mechanisms of nabilone pharmacotherapy in obesity, we did see some interesting preliminary microbiome results. Bray–Curtis dissimilarity and Aitchison distance are beta diversity metrics measuring the dissimilarity between two microbial communities. Although both beta diversity metrics are commonly used, the Aitchison distance accounts for the compositional nature of microbiome sequencing data. 30 The Bray–Curtis dissimilarity, within each individual between baseline and Week 11, was higher in the low dose nabilone group than the placebo group, providing some evidence of overall changes in gut microbiome composition based on medication group. However, this trend was not seen for the Aitchison distance. Despite the low sample size, these data suggest that nabilone treatment may indeed alter the gut microbiome relative to placebo. Due to the strong correlation between weight loss and medication group, we were unable to determine if weight loss or the nabilone itself were driving the changes in overall microbial composition. Further, in a previous study, it was shown that changes in the fecal microbiome composition of mice with diet-induced obesity can be mitigated with chronic exposure to THC, 18 emphasizing the importance of further studies on the relationship between CB1 agonists, obesity, and the gut microbiome.
Taken together, this pilot trial provided useful information and insights into cannabinoid pharmacotherapy in patients with obesity. It seems that a low dose nabilone could be better tolerated than a higher dose. We also hypothesize that a slower titration schedule may be needed for better tolerability. Other potential CB1 receptor ligands, such as peripherally restricted agonists/antagonists and receptor negative allosteric modulators have shown promise for treating obesity 6 and may have better tolerability. Finally, compounds targeting other components of the ECS (e.g., enzymes involved in the synthesis and degradation of the endocannabinoids, such as fatty acid amide hydrolase and monoacylglycerol lipase) may have anti-obesity effects without directly targeting the CB1 receptor. 13
Conclusion
Our trial found generally poor tolerability of nabilone treatment for obesity in adults, especially at the higher dose (6 mg/day). The low nabilone dose was associated with reductions in body weight and BMI that were not seen in the placebo group, though our results should be taken with caution with the limited sample size. Preliminary analysis suggested that treatment with nabilone may alter the gut microbiome relative to placebo, though this requires replication in a properly powered trial. Further studies are required to test the possibility utility of ECS drugs for obesity treatment.
Footnotes
Acknowledgment
The authors thank Dr. Alain Dagher and Dr. Sean Wharton for their guidance on this project. The authors also thank Zoe Bourgault for assisting with the trial.
Authors’ Contributions
J.M.: Conceptualization, formal analysis, investigation, data curation, writing—original draft, writing—review and editing, and visualization. D.T.: Formal analysis, data curation, writing—original draft, writing—review and editing, and visualization. S.M.: Conceptualization, methodology, writing—review and editing, and funding acquisition. V.T.: Writing—review and editing, and funding acquisition. S.C.: Writing—review and editing. K.A.S.: Conceptualization, writing—review and editing, and funding acquisition. C.S.: Conceptualization, methodology, and writing—review and editing. M.S.: Conceptualization; methodology, writing—review and editing, supervision, funding acquisition. B.L.F.: Conceptualization, methodology, writing—review and editing, supervision, and funding acquisition.
Author Disclosure Statement
B.L.F. has obtained funding from Indivior for a clinical trial sponsored by Indivior. B.L.F. has in-kind donations of placebo edibles from Indiva. B.L.F. has obtained industry funding from Canopy Growth Corporation (through research grants handled by the CAMH and the University of Toronto). He has participated in a session of a National Advisory Board Meeting (Emerging Trends BUP-XR) for Indivior Canada and is part of Steering Board for a clinical trial for Indivior. He has been consultant for Shinogi and ThirdBridge. He got travel support to attend an event by Bioprojet. He is member of the Scientific Advisory Board for NFL Biosciences. He is supported by CAMH, Waypoint Center for Mental Health Care, a clinician-scientist award from the Department of Family and Community Medicine of the University of Toronto and a Chair in Addiction Psychiatry from the Department of Psychiatry of University of Toronto. No other authors have anything relevant to declare.
Funding Information
This work was funded by a Grant # 18-12 from the Physicians’ Services Incorporated Foundation.