
Abstract
Objectives:
This study evaluated flexible-dose pharmacokinetics, safety, and effectiveness of aripiprazole in children and adolescents with conduct disorder (CD).
Methods:
This open-label, 15-day, three-center study with an optional 36-month extension enrolled a total of 23 patients: 12 children (6–12 years) and 11 adolescents (13–17 years) with CD and a score of 2–3 on the Rating of Aggression Against People and/or Property (RAAPP). Initially, the protocol used the following dosing: subjects <25 kg, 2 mg/day; subjects 25–50 kg, 5 mg/day; subjects >50–70 kg, 10 mg/day; and subjects >70 kg, 15 mg/day. Due to vomiting and sedation, this schedule was revised to: <25 kg, 1 mg/day; 25–50 kg, 2 mg/day; >50–70 kg, 5 mg/day; and >70 kg, 10 mg/day.
Results:
Aripiprazole pharmacokinetics were linear, and steady state appeared to be attained within 14 days. Both groups demonstrated improvements in RAAPP scores and Clinical Global Impressions–Severity (CGI-S) scores. Adverse events were similar to the known profile for aripiprazole in adults.
Conclusion:
The pharmacokinetics of aripiprazole in children and adolescents are linear and comparable with those in adults. Aripiprazole was generally well-tolerated in patients with CD, particularly after protocol adjustments, with improvements in aggressive behavior.
Introduction
Aripiprazole is now indicated in the United States for the treatment of schizophrenia and bipolar disorder in both adult and adolescent patients. At the time that this study was conducted, however, there were no data (pharmacokinetic or clinical) available on aripiprazole in children or adolescents. The pharmacokinetic profile of aripiprazole in healthy adults has been found to be linear up to a dose of 30 mg/day, with an oral bioavailability of 87% and a mean elimination half-life of 47–68 hours (Mallikaarjun et al. 2004; Abilify 2007). Studies in adults indicate that aripiprazole, within the dose range of 10–30 mg/day, has efficacy and an acceptable safety profile in a variety of psychotic and mood disorders (Kane et al. 2002; Kasper et al. 2003; Keck et al. 2003; Marder et al. 2003; Pigott et al. 2003; Potkin et al. 2003; Andrezina et al. 2006; Keck et al. 2006; Berman et al. 2007; Zimbroff et al. 2007; Marcus et al. 2008), including symptoms of hostility in schizophrenia patients (Volavka et al. 2005) or agitation in patients with schizophrenia, schizoaffective disorder, or bipolar disorder (Andrezina et al. 2006; Zimbroff et al. 2007). Given the clinical safety and tolerability of aripiprazole in adults, as well as its distinct pharmacologic profile (Burris et al. 2002; Jordan et al. 2002; Shapiro et al. 2003; Stark et al. 2007), aripiprazole was considered to be an appropriate medication to investigate in pediatric patients. As a first step in this exploration, pharmacokinetic/dosing evaluations were required to establish appropriate dosing paradigms for aripiprazole in children and adolescents.
Conduct disorder (CD) is a condition that is characterized, in part, by inappropriately aggressive behavior toward people and property and is associated with an increased risk of antisocial behavior in adulthood (American Psychiatric Association 1994). There is a body of evidence to suggest that treatment with an antipsychotic agent (either typical or atypical) may be a rational management option for some youngsters suffering from CD or its related conditions (Findling et al. 2008 ), because aggression is a common target symptom for antipsychotic pharmacotherapy in the young (Steiner 2003). Despite the data that are available to suggest that treatment with an antipsychotic agent might be associated with symptom amelioration in this patient population, concerns about the safety of these drugs in children remain (Bryden et al. 2001; Findling 2005). This study aimed to determine the flexible-dose pharmacokinetics, safety, and effectiveness of aripiprazole in children and adolescents with CD.
Methods
This study was conducted in accordance with Good Clinical Practice procedures, Food and Drug Administration regulations, and the Declaration of Helsinki. The design was approved by the Institutional Review Board at each participating site. For each patient, a parent/legal guardian gave informed written permission for participation in the study. Subjects provided written, informed assent, as developmentally appropriate.
Study design and subjects
This was an open-label study consisting of two phases. Phase A was a 15-day, outpatient pharmacokinetic study. Prior to dosing on days 1 and 14, subjects were brought to the investigator's center so that 24-hour postdosing assessments could be completed. The subjects stayed at the investigator's center for 24 hours to collect blood samples for pharmacokinetic analyses. On those days, doses were administered by study personnel. In addition, subjects were to report to the clinical facility for outpatient visits on the morning on days 7 and 10. On days when subjects did not have scheduled study visits, they were to take their daily dose of aripiprazole at home. On these days, caregivers were asked to note the dosing time in a diary. Phase B was a 36-month safety extension. During Phase B, subjects were to visit the study sites during months 1, 2, 3, 4, 5, 6, 9, 12, 15, 18, 21, 24, 27, 30, 33, and 36 for safety and pharmacodynamic assessments and possible dose adjustment.
Children and adolescents aged 6–12 years and adolescents aged 13–17 years were recruited from three centers and were required to have a confirmed primary diagnosis of CD (as defined by Diagnostic and Statistical Manual of Mental Disorders, 4th edition [DSM-IV] criteria) (American Psychiatric Association 1994) (with or without co-morbid attention-deficit/hyperactivity disorder [ADHD] [DSM-IV criteria]). Each prospective subject underwent a diagnostic assessment by a trained interviewer using the Schedule for Affective Disorders and Schizophrenia for School Age Children: Present and Lifetime Version (K-SADS-PL) (Kaufman et al. 1997), as well as a clinical interview with a psychiatrist. In addition, subjects had to score a 2 or a 3 on the Rating of Aggression Against People and/or Property (RAAPP) Scale (Kemph et al. 1993), have an intelligence quotient (IQ) of ≥70 by the Wechsler Intelligence Scale for Children, Version III (Wechsler 1991), within 6 months prior to enrollment (12 months for subjects aged 13–17 years), and be otherwise healthy. Subjects were excluded if they had any other major psychiatric or neurological disorder (except ADHD), had a history of recent drug or alcohol abuse (DSM-IV criteria) and/or a positive urine screen for drugs of abuse, were below the 25th percentile for their age in body weight and height, or were pregnant or lactating.
Dosing
Initially, a dose range of aripiprazole 2–15 mg/day (approximately 0.2 mg/kg aripiprazole) was selected for this study based on a review of the clinical experience at that time with aripiprazole in adults. Doses were determined by body weight, as shown in Table 1. Four out of the first 5 subjects (1 adolescent weighing 132 kg and 4 children weighing 23–41 kg) enrolled experienced tolerability issues, namely vomiting and sedation. Due to these findings, the dosing for this study was changed. Subsequently, during Phase A (screening to day 15), subjects were to receive daily doses of approximately 0.1 mg/kg aripiprazole starting on day 1 and continuing through day 14. Specifically, subjects weighing less than 25 kg were to receive 1 mg, subjects weighing between 25 and 50 kg were to receive 2 mg, subjects weighing >50 and 70 kg were to receive 5 mg, and subjects weighing more than 70 kg were to receive 10 mg. If doses of 2–10 mg aripiprazole were poorly tolerated, subsequent doses could be reduced at the investigator's discretion (see Table 1). Subjects received their allocated dose from day 1 through to day 14, administered once in the morning.
Dosing revised following tolerability issues (vomiting and somnolence).
Following discharge from the clinic on day 15 at the end of Phase A, subjects were permitted to enter the open-label extension period (Phase B) with no interruption of dosing. Throughout Phase B, the dose could be adjusted at the discretion of the investigator up to a maximum of 15 mg/day.
All 5 subjects who began on the initial regimen completed their participation in Phase A of the study. The adolescent who started on 15 mg continued on this dose throughout Phase A. Two of the children who had tolerability issues did not have their dose changed (2 mg or 5 mg) despite their initial adverse events (AEs). The remaining 2 children had their doses reduced from 5 mg (first dose) to 2 mg for the rest of Phase A. Two of these 4 children discontinued later during Phase B on days 71 and 75 because they were lost to follow up.
Assessments
Pharmacokinetic assessments
Blood samples were taken prior to dosing and at 1, 2, 3, 4, 6, 8, 12, and 24 hours postadministration on days 1 and 14. Additional samples were collected prior to dosing on days 7 and 10 to determine trough (Cmin) aripiprazole concentrations. Plasma samples were assayed for concentrations of aripiprazole and dehydroaripiprazole using a validated liquid chromatography-tandem mass spectroscopy (LC/MS/MS) method that was based on a previously described method (Kubo et al. 2005).
Plasma concentration versus time data for aripiprazole and its metabolite dehydroaripiprazole were analyzed by noncompartmental methods using the program MENU/PKMENU. The following pharmacokinetic values were determined for aripiprazole on days 1 and 14 of the study: Maximum plasma concentration (Cmax), time to maximum plasma concentration (Tmax), area under the plasma concentration versus time curve for the interval to time “T” (AUC(0-T)) (day 1), area under the plasma concentration versus time curve for the dosing interval (AUC(TAU)) (day 14), and apparent oral total body clearance (CLT/F) (day 14). The following pharmacokinetic values were determined for dehydroaripiprazole on days 1 and 14 of the study: Cmax, Tmax, and AUC(0-T). The AUC values were not to be reported if there were less than three measurable concentrations in the plasma concentration–time profile. In addition, trough (Cmin) aripiprazole and metabolite concentrations were recorded on days 7 and 10 of the study.
Pharmacodynamic assessments
Symptoms of aggression were assessed by investigators on days 1, 7 and 14, and months 1, 2, 3, 4, 5, 6, 9, 12, 15, 18, 21, 24, 27, 30, 33, and 36 using the following rating scales: RAAPP, Clinical Global Impressions–Severity of Illness (CGI-S), and Clinical Global Impressions–Improvement (CGI-I) (rated on days 7 and 14 only) (Guy 1976). The RAAPP is a single-item 5-point scale ranging from no aggressiveness (1) to intolerable behavior (5) (Kemph et al. 1993). CGI-S is rated on a 7-point scale ranging from normal (1) to extremely ill (7). CGI-I rates the change in symptoms over a treatment period on a scale from “very much improved” (1) through “no change” (4) to “very much worse” (7). The following tests were conducted: A Neuropsychological Test Battery (Wisconsin Card Sort Test, pediatric version [WCST]) total mean errors; (Berg 1948); Conners' Continuous Performance Test (Connors and Staff 2000); Conners' Continuous Performance Test II ([CPT II]); mean hit reaction time (Computer programs for Windows™ technical guide and software manual, North Tonawanda, NY; Multi-health Systems Inc.), Verbal Fluency Test (VFT) (Lezak et al. 1995). The number of words beginning with F, A, or S that the subject could name in 1 minute was also conducted to assess for the presence of any deleterious effects on cognitive functioning at 6 months.
Safety
Assessments of drug safety during the study included recording of spontaneously reported AEs (days 1, 2, 3–6, 7, 8/9, 10, 11–13, 14 and 15, and months 1, 2, 3, 4, 5, 6, 9, 12, 15, 18, 21, 24, 27, 30, 33, and 36); assessments of extrapyramidal symptoms (EPS) using the Simpson–Angus Scale (SAS), Barnes Akathisia Rating Scale (BARS), and the Abnormal Involuntary Movement Scale (AIMS) (days 1, 7, and 14, and months 1, 2, 3, 4, 5, 6, 9, 12, 15, 18, 21, 24, 27, 30, 33, and 36); electrocardiogram (ECG) (days 1, 7, and 14; months 1, 2, 3, 4, 5, 6, 9, 12, 15, 18); vital signs (days 1, 7, and 14; months 1, 2, 3, 4, 5, 6, 9, 12, 15, 18); and laboratory parameters (including combined fasting and nonfasting cholesterol and nonfasting glucose levels) (days 1 and 15; months 1, 2, 3, 4, 5, 6, 9, 12, 15, 18, 21, 24, 27, 30, 33, and 36).
Concomitant medication
All psychotropic medications other than the study drug were prohibited during Phase A of the study. Other psychotropic medications had to be discontinued at least 30 days before the start of aripiprazole therapy with the exception of methylphenidate and other stimulants, which had to be discontinued 1 week before aripiprazole treatment. The following additional concomitant medications were acceptable during Phase B: Antibiotics, proton pump inhibitors (e.g., omeprazole); histamine H2 receptor inhibitors (e.g., cimetidine), and antacids; antihistamines; stimulants, with the exception of pemoline, were allowed from months 2 through 36; buspirone was allowed from months 2 through 36; albuterol and inhaled steroids, as well as some nonprescription (over-the-counter) medications not specifically prohibited, were permitted throughout the study. In addition, behavioral and cognitive therapies were permitted during the study, with the following restrictions: Therapy was to be initiated more than 7 days before the next scheduled CGI, RAAPP, or neuropsychological assessment, and cognitive/behavioral interventions and sessions were not to take place within 24 hours prior to CGI, RAAPP, or neuropsychological assessments.
Statistics
All assessments and analyses were carried out on the children and adolescents as two separate subgroups because of developmental differences between these groups. Statistical analysis was carried out using SAS/STAT®, version 8.2. Only patients with a baseline visit and at least one other postbaseline visit were included in the analyses. Summary statistics (mean and median [standard deviation, SD]) for the pharmacokinetic parameters of aripiprazole and dehydroaripiprazole were calculated by study day and aripiprazole dose for the patient population. Frequency distributions and summary statistics of the RAAPP, the CGI variables, count data from the neuropsychological test battery (WCST, CPT, and VFT), and corresponding changes from baseline (observed cases, OC) were tabulated.
Results
Demographics
All 23 subjects (12 children aged 6–12 years, and 11 adolescents aged 13–17 years) who entered the study completed the initial 14 days of treatment (Phase A) and continued into the 36-month continuation phase (Phase B). Of these, 5 patients (21.7%, 2 children and 3 adolescents) completed 36 months of treatment and 18 (78.3%) discontinued prior to the 36-month time point. The reasons for discontinuation were lost to follow up (7/23 = 30.4%), nonadherence with study-related procedures (6/23 = 26.1%), withdrawal of consent (3/23 = 13.0%), and lack of efficacy (2/23 = 8.7%).
Demographic characteristics of the treated patients were as follows: Mean ± SD age of the children was 8 ± 2 years and of the adolescents was 14 ± 1 years. The distribution of male and female patients among the children was 8 males and 4 females and among the adolescents was 9 males and 2 females. Among the children, 5 were white and 7 were black; among the adolescents, 8 were white and 2 were black. The children had a mean weight of 36.3 ± 14.5 kg and height of 131.4 ± 11.3 cm, whereas the adolescents were 73.0 ± 28.5 kg and 163.7 ±8.5 cm in height. Pharmacodynamic data collected after day 1 were excluded for 1 patient due to noncompliance with study-related procedures.
Dosing
At the end of Phase A, patients were receiving final doses of aripiprazole between 1 mg/day and 15 mg/day. In this phase, 4 patients received the maximum dose of 10 mg/day or 15 mg/day. Aripiprazole dose was adjusted upward in Phase B for most patients (n = 14/23, 60.9%), with 6 of these 14 patients eventually receiving the maximum dose. With the exception of 1 patient, all patients received aripiprazole for more than 30 days. The mean duration of exposure to the study treatment was 393 days for children and 382 days for adolescents.
Pharmacokinetics
The plasma concentration-time curves on Day 1 and Day 14 for aripiprazole in children and adolescents are shown in Fig. 1. The pharmacokinetic parameter estimates for each aripiprazole dose in the two age groups are summarized in Table 2. Upon visual inspection, steady-state appears to have been attained within 14 days of once-daily aripiprazole dosing. Comparison of Cmax and AUC values for day 1 and day 14 throughout the study indicated that day-14 values were two- to four-fold higher for Cmax and three- to six-fold higher for AUC than the values observed on day 1.
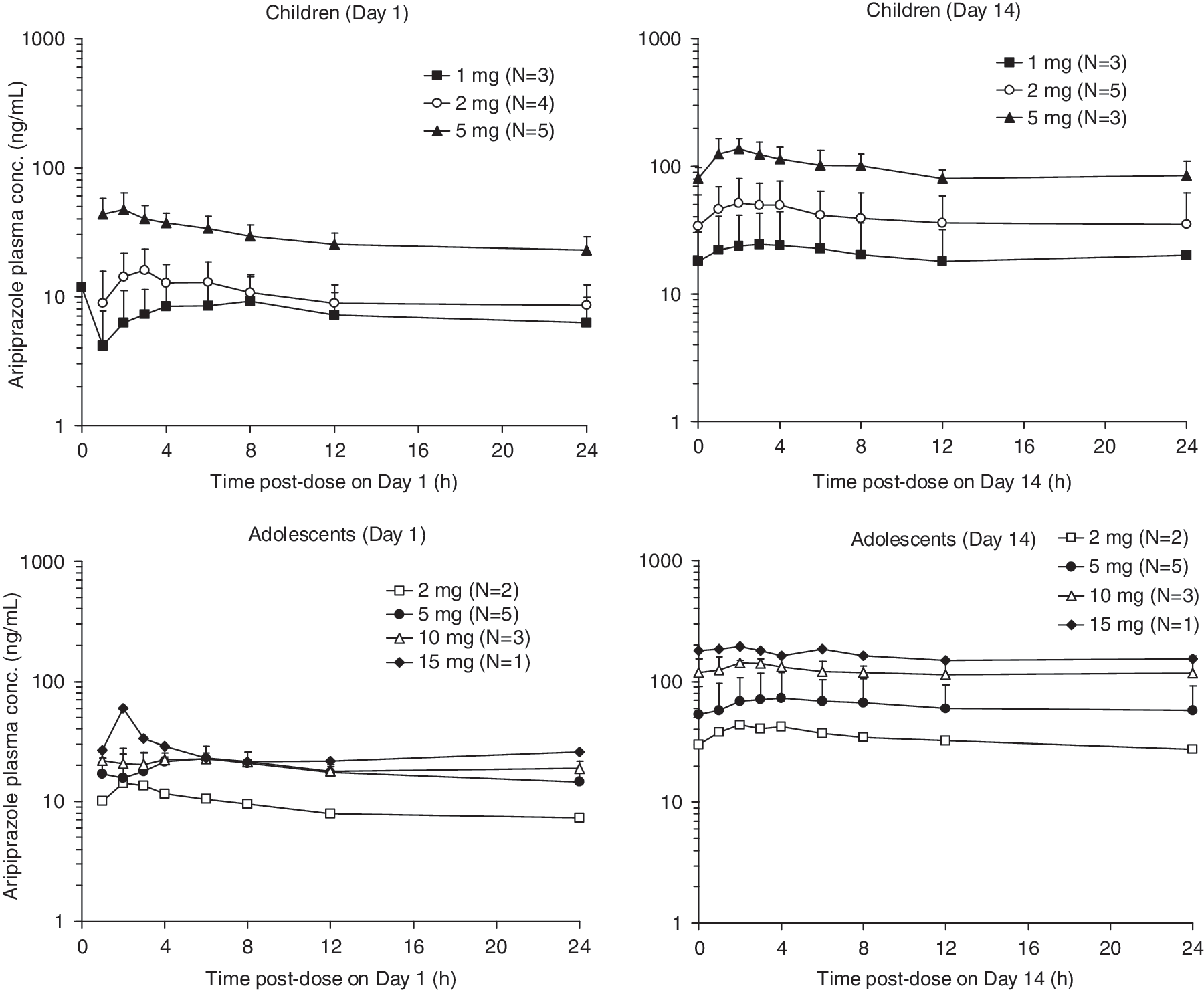
Plasma concentration–time profiles (mean ± SD) of aripiprazole in children (6–12 years) and adolescents (13–17 years). Notes: Values less than the lower limit of quantitation (0.5 ng/mL) were treated as missing for the calculation of mean ± standard deviation (SD) values; error bars are not shown for ≤2 subjects.
aRepresents AUC(0-T) on Day 1 and AUCt on Day 14; b n = 2; cnot reported because n = 2; d n = 1.
These n numbers represent Day 14; at Day 1 n = 2 with 1 mg, n = 4 with 2 mg, and n = 5 with 5 mg.
Abbreviations: Cmax = maximum plasma concentration; N/A = not applicable (n = 1); CV = coefficient of variation; CLT/F = apparent oral total body clearance; SD = standard deviation; Tmax = time to maximum plasma concentration; AUC = area under the curve; GEO = geometric.
Comparison of Cmax and AUC values for day 14 for the different doses showed that both parameters appeared to be linearly proportional to the administered dose in both age groups. The mean apparent oral clearance of aripiprazole, unadjusted for body weight, was slightly lower for children than adolescents (2.53 ± 1.06 L/hour vs. 3.85 ± 1.46 L/hour). However, when normalized for body weight, the values for the mean apparent oral clearance were similar across age groups.
Pharmacokinetic parameters were also evaluated for the major metabolite of aripiprazole, OPC-14857, which has been shown to have a similar affinity for D2 receptors as aripiprazole. The pharmacokinetic parameters for this active metabolite at each aripiprazole dose in the two age groups are also summarized in Table 2.
Effectiveness
On day 1, prior to dosing, the RAAPP scores (mean ± SD) were 3.00 ± 0.63 for children (median 3) and 2.64 ± 0.50 for adolescents (median 3). Overall, RAAPP scores improved during the course of the study. Both children and adolescents showed a median score of 2 at day 14 and remained at this level at month 36. Similar decreases in RAAPP scores (mean ± SD) from baseline were seen both in children (day 14, −0.82 ± 0.75; month 36, −1.00 ± 1.00) and in adolescents (day 14, −1.00 ± 0.89; month 36, −0.75 ± 0.96).
On day 1, the CGI-S scores (mean ± SD) were 4.27 ± 1.01 for children (median 4) and 3.82 ± 0.98 for adolescents (median 4). CGI-S scores showed improvements at day 14 and month 36 for both age groups. For children, the median score decreased from 4 (moderately ill) at day 1, to 3 (mildly ill) at day 14, to 2 (borderline ill) at month 36. In adolescents, the median score decreased from 4 (moderately ill) at day 1, to 2 (borderline ill) at day 14, and remained at this level to month 36. The frequency distribution of CGI-I scores confirmed the clinical improvement seen with aripiprazole. By day 14, 63.6% of children and 45.5% of adolescents were rated as much or very much improved on the CGI-I score. At month 36, 66.7% of children and 100% of adolescents showed this level of CGI-I score (much or very much improved) (OC).
The neuropsychological battery showed, on average, minor improvements. On the WCST, the ability to sort cards according to color, shape, and number categories improved in both children and adolescents over 6 months, with the mean (SD) total errors in children decreasing from 54 (17) at screening to 31 (32) at month 6 and in adolescents from 28 (20) at screening to 11 (6) at month 6. Similarly, minor improvements were seen in the mean (SD) hit reaction time on the CPT in both groups, with a decrease in children from 507 (74) msec at screening to 470 (60) msec at month 6 and in adolescents from 390 (70) msec at screening to 367 (89) msec at month 6. Third, the VFT showed that patients increased the number of words they could generate in 1 minute by no more than 2 at any of the time points assessed.
Safety/tolerability
Treatment with aripiprazole was generally well tolerated after the dose adjustment (Table 3) that was implemented following vomiting in 4 and somnolence in 3 children. Throughout the study, there were 5 reports of EPS, 3 of them being in Phase A. In Phase A, EPS were reported in 2 children (cogwheel rigidity, n = 1 [8.3%]; and tremor, n = 1 [8.3%]), and reported by 1 adolescent (hypokinesia, 9.1%). In Phase B, one child reported tremor (8.3%) and 1 adolescent reported essential tremor (9.1%). All EPS were considered to be of mild intensity by the investigator and did not lead to discontinuation. The cogwheel rigidity and hypokinesia noted in Phase A resolved with a dose reduction. The tremors (1 each in Phases A and B) resolved with no action taken. The essential tremor in Phase B did not resolve, but it was considered to be unrelated to the study drug by the investigator and no action was taken. No patient scored higher than “2” on any item at any time point on the SAS; all patients scored “0” (no awareness) on AIMS items 1–7 at all time points; and in the BARS (Item 4), only 1 13-year-old patient scored “1” (presence of restless movements) at month 1 (baseline). This patient scored “0” at all other evaluation points. All other subjects scored “0” (normal) at all time points.
In Phase A, dose reductions occurred as a result of vomiting (n = 4) and somnolence (n = 3) in children.
There were no serious AEs, no subjects discontinued due to AEs, and there were no deaths in either Phase A or Phase B of this study. Only two laboratory findings of potential clinical significance were observed. First, 1 patient had an elevated creatine phosphokinase on day 737 that lasted for 3 days; the investigator considered this to be not clinically significant and was not reported as an AE. Second, 1 patient had a mild elevation of hepatic enzymes on day 169 that lasted for 15 days (aspartate aminotransferase of 185 U/L [normal range 6–44 U/L] and an elevated alanine aminotransferase of 236 U/L [normal range 5–47 U/L]); the investigator considered this to be not clinically significant and was not reported as an AE. Both analytes returned to the normal range by day 183.
Elevated prolactin levels were not reported for any subjects. One patient had elevated prolactin levels at baseline, which decreased (from 28 ng/mL to <0.6 ng/mL [normal values for the laboratory used in this study ranged from 0 to 20 ng/mL]) over the study duration. In patients ≤12 years old, the mean change in prolactin levels (last observation carried forward, LOCF) from baseline (7.3 ± 3.7 ng/mL) was −5.5 ± 4.3 ng/mL. In patients ≥13 years old, the mean change in prolactin levels from baseline (9.0 ± 3.7 ng/mL) to week 72 was −2.2 ± 4.5 ng/mL.
Seventeen of the 23 subjects (73.9%) experienced vital sign abnormalities during this study. None of these vital sign abnormalities was considered to be clinically significant by the investigators. No subjects were discontinued from the study because of vital sign abnormalities. Mean weight change (LOCF) in patients ≤12 years old from baseline (36.6 ± 14.4 kg [OC, 25.2 ± 1.9 kg]) to week 72 was 9.0 ± 11.0 kg (OC, 24.1 ±12.1). In patients ≥13 years old, the mean change in weight (LOCF) from baseline (73.3 ± 28.8 kg (OC, 91.7 ± 21.4)) was 13.3 ± 15.5 kg (34.4 ± 15.3). Mean BMI change (LOCF) in patients ≤12 years old from baseline (21.6 ± 6.1 kg/m2 [OC, 16.7 ± 1.3 kg/m2]) to week 72 was 1.8 ± 4.2 kg/m2 (OC, 6.9 ± 5.2 kg/m2). In patients ≥13 years old, the mean change in BMI (LOCF) from baseline (27.1 ± 10.0 kg/m2 [OC, 33.9 ± 3.3 kg/m2]) was 3.4 ± 4.7 kg/m2 (OC, 9.3 ± 6.1 kg/m2). No patients discontinued due to weight gain. Total cholesterol level (OC) in patients ≤12 years old at week 2 was 151.3 mg/dL (baseline 144.0), at week 12 was 164.9 mg/dL (baseline 149.8) and at week 48 was 156.0 mg/dL (baseline 149.0). In patients ≥13 years old, cholesterol level (OC) at week 2 was 163.6 mg/dL (baseline 164.7), at week 12 was 161.7 mg/dL (baseline 164.5) and at week 48 was 168.5 mg/dL (baseline 172.8). Glucose level (OC) in patients ≤12 years old at week 2 was 92.7 mg/dL (baseline 88.8), at week 12 was 88.6 mg/dL (baseline 88.2) and at week 48 was 81.5 mg/dL (baseline 99.0). In patients ≥13 years old, glucose level (OC) at week 2 was 100.9 mg/dL (baseline 97.1), at week 12 was 88.8 mg/dL (baseline 97.1), and at week 48 was 104.7 mg/dL (baseline 110.0). This OC data is corroborated by LOCF findings (data not shown).
Discussion
Given the established efficacy and tolerability of aripiprazole in adults with psychiatric disorders (Kane et al. 2002; Kasper et al. 2003; Keck et al. 2003; Pigott et al. 2003; Volavka et al. 2005; Andrezina et al. 2006; Keck et al. 2006; Berman et al. 2007; Keck et al. 2007; Zimbroff et al. 2007; Marcus et al. 2008), this pilot study sought to determine, for the first time, the flexible-dose pharmacokinetics, safety, and effectiveness of aripiprazole in children and adolescents.
Following the occurrence of vomiting and sedation in 4 of the 5 patients who began treatment, the protocol was amended to decrease the dose from 2–15 mg/day to 1–10 mg/day. The tolerability issues did not continue subsequent to the amended regimen and all patients who started at the initial schedule continued in the study. During the 36-month continuation phase, the aripiprazole dose was adjusted upward in most patients.
With the implementation of a revised starting dose paradigm, aripiprazole appeared to be generally safe and well tolerated in children and adolescents with CD over the short and long term. There were no deaths or other serious AEs and no subjects were discontinued due to AEs in either Phase A or B. The AE profiles were generally similar in children and adolescents, with most of the AEs occurring in the gastrointestinal and nervous systems. The pattern of AEs in the two phases suggests that the dosing regimen used in the study resulted in improvements in long-term tolerability (for example, less vomiting and sedation) in children and adolescents. The occurrence of EPS-related AEs was low. In addition, the EPS-related AEs were of mild intensity and did not lead to discontinuation, confirming the tolerability of aripiprazole in this population. Although weight and BMI increased in children and adolescents over the study duration, weight gain in this population is normal and the findings are, therefore, not unexpected. It is important to note that no patient discontinued due to weight gain.
This study showed that the pharmacokinetics of aripiprazole in children and adolescents are linear and comparable with those in adults. The body-weight-adjusted CL/F of orally administered aripiprazole was similar in children and adolescents and comparable to that previously reported for adult patients with schizophrenia or schizoaffective disorder. The extent of accumulation of aripiprazole on day 14 compared to day 1 was similar for children and adolescents, and was consistent with the extent of accumulation observed in adults, as were the ratio of parent drug:metabolite (Mallikaarjun et al. 2004). Steady state appeared to be attained within 14 days of once-daily aripiprazole dosing in both children and adolescents, similar to the time to steady state in adults.
The study presented herein is of clinical relevance because it was the first aripiprazole study conducted in children; it was carried out before aripiprazole was commercially available as part of the clinical research program, and it provided valuable information with regard to appropriate starting dose and inferences about dose escalation that informed subsequent studies.
An additional pharmacokinetic study that was subsequently conducted has recently been published. That study confirmed the linearity of aripiprazole pharmacokinetics up to the maximum approved adult dose (30 mg/day) in children and adolescents (aged 10–17 years) with primary psychiatric diagnoses of a bipolar or schizophrenia spectrum disorder in a multicenter, open-label, sequential-cohort, dose-escalation study. Aripiprazole treatment was generally well tolerated in that study as well (Findling et al. 2008 ).
This study also provided the opportunity to assess the potential effectiveness of aripiprazole in youths with CD. Preliminary data suggest that aripiprazole may improve symptoms of CD with modest impact on cognitive function in both children and adolescents; however, the sample size is too small to draw any firm conclusions. Although this pilot study provided important pharmacokinetic information in children and adolescents that guided dosing of aripiprazole in subsequent clinical trials, interpretation of the safety and effectiveness data are limited due to the small sample size, absence of a control treatment arm, and open-label design.
This clinical trial is an example of why methodologically stringent dosing and pharmacokinetics should be performed in children and adolescents. The initial dosing regimen in this study was based on weight-adjusted dosing data in adults. However, it was subsequently observed that lower starting doses of this agent were necessary in juveniles due to vomiting and sedation. Furthermore, optimal therapeutic benefit for most patients was achieved with subsequent increases from initial doses. Thus, this study provided evidence that to treat these youths with aripiprazole optimally, smaller initial starting doses than might be inferred from adult data are necessary. In addition, despite the fact that vomiting and sedation were seen with higher starting doses, once an acceptable starting dose is identified, subsequent dose increases may be both tolerable and necessary to achieve optimal therapeutic benefit.
In conclusion, this study provides information regarding the dosing, pharmacokinetic profile, tolerability, and effectiveness of aripiprazole in pediatric patients with CD. Aripiprazole appeared to be safe and generally well tolerated by pediatric patients when appropriate dosing was used.
Footnotes
Disclosures
Dr. Findling receives or has received research support, acted as a consultant, and/or served on a speaker's bureau for Abbott, Addrenex, AstraZeneca, Bristol-Myers Squibb, Forest, GlaxoSmithKline, Johnson & Johnson, Lilly, Neuropharm, Novartis, Organon, Otsuka, Pfizer, Sanofi-Aventis, Sepracore, Shire, Solvay, Supernus Pharmaceuticals, Validus, and Wyeth. Ralph E Kauffman has received research support and/or acted as a consultant for AlgoRx Pharmaceuticals, Aventis, Boots Laboratories, Bristol-Myers Squibb, Burroughs-Wellcome, Daiichi Asubio, Eli Lilly, Hoechst-Marion Roussel, Hoffman LaRoche, Johnson & Johnson, McNeil Consumer Products, McNeil Laboratories, Ortho-McNeil, Otsuka Pharmaceuticals, Purdue Pharma, Sanofi-Synthélabo, Syntex, and Upjohn. Daniel E Salazar, Ph.D., Vaishali Sahasrabudhe, Ph.D., Georgia Kollia, Ph.D., David M Kornhauser, M.D., Nimish N Vachharajani, Ph.D., and David W Boulton, Ph.D. were current employees of Bristol-Myers Squibb at the time of this study.
Acknowledgments
Editorial support for the preparation of this manuscript was provided by Ogilvy Healthworld Medical Education and was paid for by Bristol-Myers Squibb Company.