
Abstract
Objective:
The aim of this study was to evaluate the efficacy and safety of two dose ranges of risperidone in adolescents with schizophrenia.
Methods:
In a 6-week, randomized, double-blind, placebo-controlled study, adolescents aged 13–17 years with acute exacerbation of schizophrenia were randomized to placebo, flexible doses of risperidone 1–3 mg/day, or risperidone 4–6 mg/day. Assessments included the Positive and Negative Syndrome Scale (PANSS), clinical response (≥20% reduction in PANSS total score), adverse event (AE) monitoring, and extrapyramidal symptom (EPS) scale ratings.
Results:
A total of 160 subjects received placebo (n = 54), risperidone 1–3 mg/day (n = 55), or risperidone 4–6 mg/day (n = 51). Significant improvements occurred in both risperidone groups versus placebo (p < 0.001) in PANSS total change scores (placebo, −8.9 [16.1]; risperidone 1–3 mg, −21.3 [19.6]; risperidone 4–6 mg, −21.2 [18.3]) and clinical response rates (35%, 65%, 72%, respectively). Overall AE rates were more common in risperidone groups (75% and 76%) versus placebo (54%). Risperidone 4–6 mg/day had a higher incidence of extrapyramidal disorder, dizziness, and hypertonia than risperidone 1–3 mg. No prolactin-related AEs occurred. Overall EPS severity was low.
Conclusions:
Risperidone 1–3 mg/day and 4–6 mg/day were well tolerated and effective in adolescents experiencing acute episodes of schizophrenia. The benefit–risk profile suggests that a dose of 1–3 mg/day might be optimal for this population.
Introduction
The use of antipsychotics for the treatment of schizophrenia in adult patients—specifically in reducing psychotic symptoms, preventing relapse, and improving overall functioning—is well established (Lehman et al. 2004; McClellan et al. 2007). In contrast, the definitive evidence for use of these agents in younger populations is strikingly limited, although several antipsychotics have been evaluated in small, randomized controlled studies in younger subjects with schizophrenia (Kumra et al. 1996; Sikich et al. 2004; McClellan et al. 2007; Kumra et al. 2008).
Among these evaluations, the safety and efficacy of the atypical antipsychotic risperidone have been demonstrated in children and adolescents for the treatment of disruptive behavior disorders and of irritability and other behavioral symptoms associated with autism in double-blind placebo-controlled studies (Aman et al. 2002; McCracken et al. 2002; Snyder et al. 2002; Reyes et al. 2006a; Reyes et al. 2006b; Reyes et al. 2006c). Additional experience has been obtained from noncontrolled studies with risperidone in children and adolescents with schizophrenia, preliminarily indicating that risperidone may be an effective and safe treatment for this population (Lykes and Cueva 1996; Zuddas et al. 1996; Armenteros et al. 1997; Sourander 1997; Gothelf et al. 2003). Recently, further efficacy and safety data were reported in a double-blind randomized comparison of the conventional antipsychotic molindone with olanzapine and risperidone in pediatric patients with early-onset schizophrenia or schizoaffective disorder (Sikich 2008).
This randomized, double-blind, placebo-controlled study evaluated the efficacy and safety of two dose ranges of risperidone in the treatment of schizophrenia in adolescents experiencing acute exacerbation of symptoms.
Methods
This randomized, double-blind, parallel-group, placebo-controlled, multicenter trial was conducted at 23 sites in India (58 subjects), Russia (37 subjects), Ukraine (32 subjects), and the United States (33 subjects) from August 3, 2004, to December 27, 2005. The study protocol and amendment were approved by each institutional review board or independent ethics committee prior to study initiation, and the trial was conducted in accordance with the Declaration of Helsinki.
Participants
The study included male and female subjects, aged 13–17 years, with a DSM-IV diagnosis of schizophrenia. Subjects were inpatients or outpatients; all were experiencing an acute episode with a total Positive and Negative Syndrome Scale (PANSS) score (Kay et al. 1987) of 60–120 (inclusive). Subjects were in good physical health, as determined by screening procedures, and had no other serious illnesses or neurologic conditions. Female subjects of childbearing potential were required to have a negative pregnancy test and (if sexually active) to be using an acceptable form of contraception. A responsible person was required to be available to accompany the subject at each assessment and to dispense medication accurately and reliably for subjects who were at risk for noncompliance. Participating subjects had to be willing to be hospitalized during the study if deemed clinically necessary. All subjects consented to participate in the study prior to screening, and subjects' legal representatives also provided written consent. Subjects were not compensated for their participation in the study.
Subjects who met DSM-IV criteria for dissociative disorder, bipolar disorder, major depressive disorder, schizoaffective disorder, schizophreniform disorder, autistic disorder, or primary substance-induced psychotic disorder at screening were excluded from the study. Also excluded were subjects with mild, moderate, or severe mental retardation (intelligence quotient [IQ] <70); known or suspected substance dependence diagnosed by DSM-IV criteria in the 3 months preceding screening; or significant risk of suicide or violent behavior. Subjects were excluded if they failed to respond to adequate treatment with two or more typical or atypical antipsychotics (including risperidone) during the current psychotic episode, exhibited hypersensitivity or intolerance to risperidone, or had a history of neuroleptic malignant syndrome or any severe drug allergy or hypersensitivity. Other exclusions were for depot antipsychotic treatment (within two treatment cycles before baseline), electroconvulsive therapy (in the 4 weeks before baseline), clozapine (within 2 months before baseline), and use of protocol-prohibited concomitant medications that could not be discontinued per the investigator's judgment.
Prohibited concomitant medications included antidepressants, mood stabilizers (lithium and other antimanic agents), anticonvulsants, psychostimulants and direct dopamine agonists, cholinesterase inhibitors, any herbal or over-the-counter medications with psychotropic properties, or antipsychotics other than the study medication. Drugs with sedative, hypnotic, or anxiolytic properties were not allowed, with the exception of oral lorazepam, hydroxyzine, zopiclone, or zolpidem, to control agitation and/or insomnia.
The β-adrenergic blocker propranolol was allowed for treatment-emergent akathisia; use of other β-adrenergic blockers was prohibited. Antiparkinsonian medications could be initiated after baseline for treatment-emergent extrapyramidal symptoms (EPS). Use of all rescue medications was to be kept to a minimum, and the permitted doses of certain medications (hydroxyzine and lorazepam [or diazepam or gidazepam if lorazepam was not available]) progressively decreased over the course of the study (e.g., for lorazepam, 1.5 mg/day was permitted during days 1–7, 1.0 mg/day was allowed for days 8–14, and 0.5 mg/day for days 15–42). Rescue medications were not to be administered during or 8 hours before any behavioral assessment. Subjects were not permitted to receive insight-oriented or cognitive-behavioral psychotherapy during the study; however, they could receive limited supportive psychotherapy or psychoeducation.
Study design
The study consisted of a screening phase with a washout period (if needed), followed by a 6-week double-blind treatment phase. A child psychiatrist confirmed the diagnosis of schizophrenia and all co-morbid conditions. Diagnosis of schizophrenia was confirmed by administration of selected modules of the Kiddie Schedule for Affective Disorders and Schizophrenia for School-Aged Children–Present and Lifetime (K-SADS-PL) (Kaufman et al. 1997). Training on the proper use of the K-SADS-PL was provided during investigator meetings and via Internet-based training sessions. Reliability was determined by independent review of the first K-SADS-PL completed after training. Assessments during the screening phase included a physical examination, vital signs, height and weight measurement, Tanner staging for sexual maturation, a medical/psychiatric history, and required laboratory evaluations. Subjects currently taking protocol-prohibited concomitant medications, alcohol, or other drugs of abuse entered a washout period for a maximum of 5 days.
Following the screening and washout phase, eligible subjects were randomized to one of three treatment groups for 6 weeks: Placebo, risperidone 1–3 mg/day, or risperidone 4–6 mg/day. Treatment was administered once daily in the evening, with doses titrated from the minimum within the assigned target ranges (1 mg/day for the risperidone 1- to 3-mg group or 4 mg/day for the risperidone 4- to 6-mg group) by day 7 of treatment. Further increases within the assigned dosage range (forced titration) were made by day 14 to the maximum tolerated dosage level. After day 14, subjects were to be maintained at the maximally tolerated dose for the remainder of the study; any required dose adjustments had to be within the assigned target range or the subject was withdrawn.
Doses of study medication were administered by study-site personnel for hospitalized subjects. Outpatient subjects self-administered their medication. If the investigator judged it necessary to ensure compliance, a parent or other responsible caretaker monitored and administered medications. Subjects or their parents/caregivers were to complete a subject diary each day to indicate whether study medication was taken as directed and listing all concomitant medications.
Outcome assessments
Clinicians were trained and certified to perform all assessments. To ensure consistent administration and scoring of the PANSS in this adolescent study population, the study sponsor developed and implemented PANSS administration guidelines, a training program, and a certification process for investigators. Certification of raters consisted of detailed instruction on the PANSS, including a focus on developmental adjustments. Following training, raters were tested by scoring a tape of adolescents with schizophrenia who demonstrated sufficient positive and negative symptoms. Answer sheets were graded by comparison to an expert-derived Master Score. Rater candidates were certified by adequate scoring of this interview and by demonstrating appropriate credentials and experience. Retraining occurred at least annually. Whenever possible, individual subject assessments were made at approximately the same time of day, and administered by the same clinician at all visits.
Study assessments included the PANSS, Clinical Global Impressions–Severity (CGI-S), Clinical Global Impressions–Improvement (CGI-I) (Guy 1976a), and Children's Global Assessment Scale (CGAS) (Shaffer et al. 1983). The PANSS assesses neuropsychiatric symptoms on a 30-item scale, with each item rated from 1 (absent) to 7 (extreme). The total score has a range of 30–210, with higher scores representing more severe illness. PANSS factor scores (Marder et al. 1997) for positive symptoms, negative symptoms, disorganized thoughts, uncontrolled hostility/excitement, and anxiety/depression provide assessment of discrete symptom domains. The CGI-S rating scale provides a global evaluation of the subject's condition on a 7-point scale ranging from 1 (not ill) to 7 (extremely severe). The CGI-I provides global evaluation of improvement, with anchor points ranging from 1 (very much improved) to 7 (very much worse) (Guy 1976a). The CGAS is a 100-point rating scale measuring psychological, social, and school functioning in children aged 6–17 years. Higher scores indicate better functioning overall (Shaffer et al. 1983). The PANSS and CGI-S were administered at baseline and at weeks 1 (day 8), 2 (day 15), 4 (day 29), and 6 (day 43)/end point; the CGI-I was administered at weeks 1, 2, 4, and 6/end point; and the CGAS was administered at screening and week 6/end point.
The primary efficacy measure was a baseline-to-end point change in the PANSS total score. Secondary efficacy measures included change in mean PANSS total scores at intermediate time points, change in PANSS factor scores, number and percentage of responders to risperidone treatment (predefined as ≥20% reduction from baseline in PANSS total score), baseline-to-end point change in CGI-S score, CGI-I score at end point, and screening-to-end point change in CGAS.
Safety and tolerability were assessed via adverse events (AEs), which were reported by the subject or, when appropriate, by the subject's caregiver or surrogate. Treatment-emergent AEs were defined as those with onset during the double-blind treatment phase or within 4 days of the end of treatment. Serious AEs were recorded during the study and for 30 days following study completion. EPS were evaluated using the Abnormal Involuntary Movement Scale (AIMS) (Guy 1976b), Simpson–Angus Scale (SAS) (Simpson and Angus 1970), and Barnes Akathisia Rating Scale (BARS) (Barnes 1989). The AIMS has 10 items of involuntary movement, ranging from 0 (none) to 4 (severe). A total severity score (ranging from 0 to 28) was based on seven items for facial, oral, extremity, and trunk movement; higher scores denote greater severity. The BARS global clinical rating of akathisia rates symptoms from 0 (absent) to 5 (severe), with higher scores indicating more severe akathisia. The SAS rates 10 items related to parkinsonism symptoms from 0 (normal) to 4 (extreme). The 10 items are summed and divided by 10 so that the total SAS score ranges from 0 to 4; a higher score indicates a more severe condition. EPS rating scales were administered at baseline and weeks 1, 2, 4, and 6/end point.
Other tests performed included clinical laboratory tests (hematology, comprehensive chemistry, and urinalysis) at baseline and weeks 2 and 6/end point, electrocardiograms (at screening and end point), vital signs (at baseline and weeks 1, 2, 4, and 6/end point), and body weight and height measurements (at baseline and weeks 2, 4, and 6/end point).
Data analysis
A sample size calculation was based on the median drug–placebo difference observed in two studies of risperidone in adult subjects with schizophrenia (Marder and Meibach 1994; Peuskens 1995). Assuming that the standard deviation (SD) of the change in PANSS total score from baseline to end point would be 21.5 points, approximately 50 subjects in each treatment group with an end-point assessment were needed for the study to have approximately 85% power to detect a clinically relevant difference of 13 points in the primary efficacy measure, at the 5% significance level with a two-sided test. Assuming that 5% of subjects would discontinue the study and not have an end-point assessment, it was estimated that 53 subjects would need to be randomized to each arm. Subjects were assigned to one of the three treatment groups using a 1:1:1 ratio based on a computer-generated randomization scheme. Randomization was balanced using randomly permuted blocks and was stratified by center. All double-blinded study medications were identical in appearance and were dispensed in uniform-appearing containers.
Efficacy and safety were analyzed in the intent-to-treat (ITT) population (all randomized subjects who received at least one dose of treatment). Frequencies, percentages, and descriptive statistics were used to describe demographic/clinical characteristics and efficacy and safety variables when appropriate.
Efficacy outcome variables were analyzed at each time point and at end point, utilizing the last-observation-carried-forward (LOCF) approach. End point was defined as the last nonmissing, postbaseline value during the double-blind treatment phase that fell on or before day 50. An analysis of covariance (ANCOVA) model was used to analyze mean change in PANSS total score from baseline to end point, with treatment and country as factors and the baseline PANSS total score as covariate. Estimated least squares means of the differences, the Fisher least significant difference, p values, and 95% confidence intervals (CI) were calculated for the mean change in each risperidone dosage group versus placebo.
For the primary variable, a step-down closed testing procedure that preserved the experiment-wise α-level of significance (0.05) was used in the comparisons of the treatment groups: The risperidone 4- to 6-mg group was compared with placebo, and only if statistically significant at the 5% level, the risperidone 1- to 3-mg group was then compared with placebo. For the analysis of secondary efficacy variables, statistical tests were interpreted at the 5% significance level (two-sided) and nominal unadjusted p values reported.
Analysis of PANSS total and factor scores was performed for each assessment time point using the same ANCOVA model as for the primary efficacy end point. Clinical response rate (the percentage of subjects with a ≥20% reduction in PANSS total score from baseline) was analyzed at each time point. Between-group differences were analyzed using the Cochran–Mantel–Haenszel test for general association controlling for country. ANCOVA models, analogous to those described above, were used to assess between-treatment differences on the CGI-S, CGI-I (analysis of variance [ANOVA]), and CGAS. Weight was transformed to z-scores (and percentile ranks) based on the May 30, 2000 U.S. Centers for Disease Control growth charts. The z-score indicates how many SDs away an observed value is from the expected weight, on the basis of a subject's age and sex.
To investigate the robustness of the results from the primary analysis, a preplanned analysis of change from baseline over time in PANSS total score (using observed data) was performed using a repeated-measurements model with time, treatment, and country as fixed effects and baseline PANSS total score as a covariate. A treatment-by-time interaction term was included to evaluate whether the trends over time differed between the treatment groups. An unstructured variance–covariance matrix and the Satterthwaite approximation for the denominator degrees of freedom were used.
Results
A total of 160 subjects in the ITT population were randomly assigned to receive placebo, risperidone 1–3 mg/day, or risperidone 4–6 mg/day (Fig. 1). Efficacy data were not collected for 2 subjects (1 in each of the two active-treatment groups); efficacy analyses, therefore, included 158 subjects. Baseline demographic and clinical characteristics were generally comparable in the three treatment groups (Table 1). The median age for all groups was 16.0 years, and the majority of subjects (64%) were male. The most common diagnosis was paranoid schizophrenia (69%), and approximately half of study participants were inpatients at screening (Table 1). Overall, 40% of subjects had co-morbid psychiatric disorders (most frequently insomnia), and 4% had a neurologic condition, predominantly headaches. Five percent had a history of other co-morbid psychiatric disorders. Mean (SD) PANSS total scores at baseline were similar across treatment groups and reflected an acutely ill population: Placebo, 93.2 (10.3); risperidone 1–3 mg, 95.4 (11.0); and risperidone 4–6 mg, 93.0 (11.9). Mean (SD) PANSS total scores at baseline for hospitalized patients were 96.7 (9.3), 97.4 (11.2), and 97.6 (12.5) in the placebo, risperidone 1–3 mg, and risperidone 4–6 mg groups, respectively. As anticipated, PANSS total scores were somewhat lower in subjects who were not hospitalized: 90.6 (10.3), 93.1 (10.6), and 88.8 (9.7), respectively.
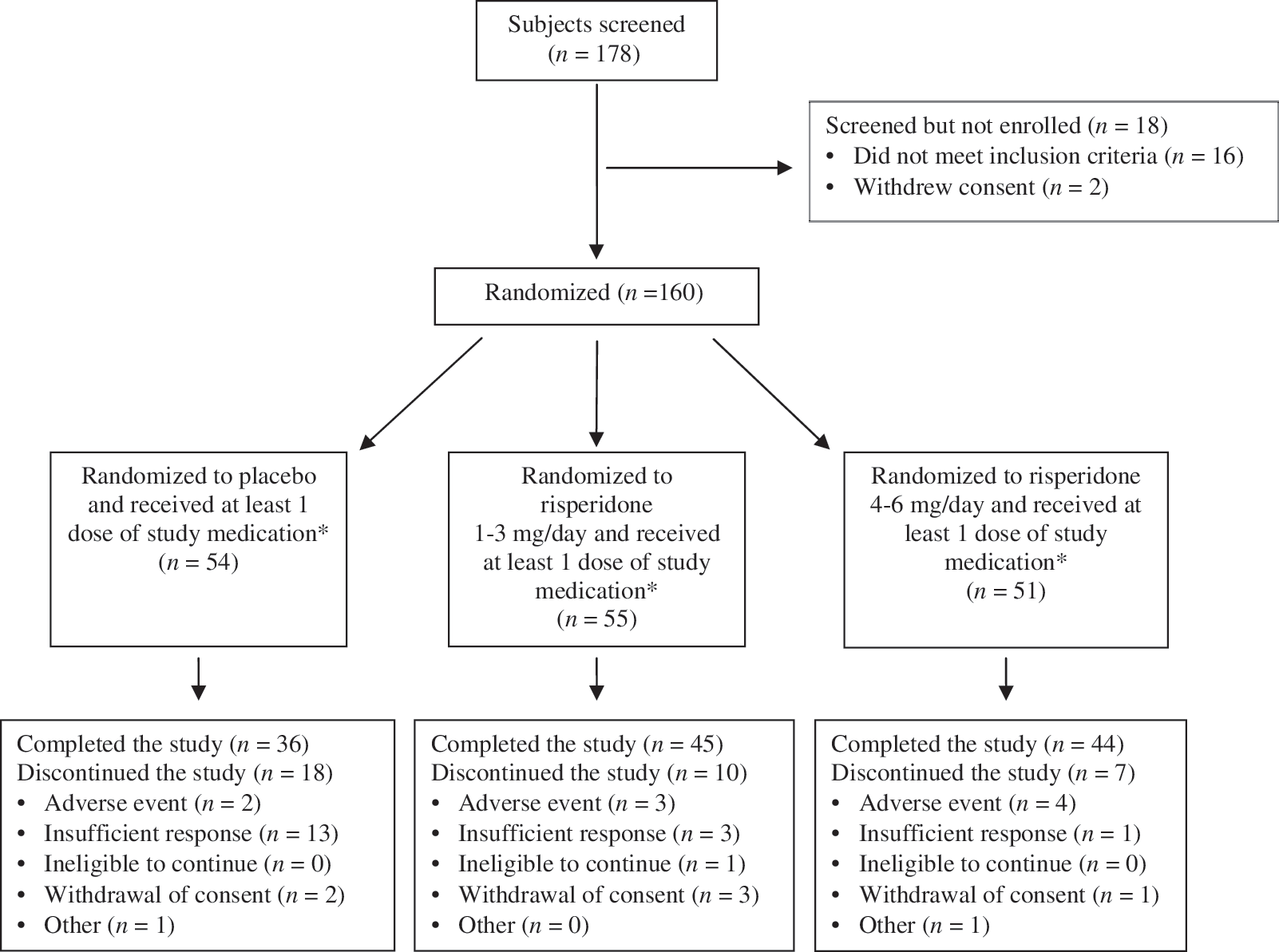
Subject flow diagram. The asterisk (*) indicates subjects in the intent-to-treat (ITT) population, which included all randomized subjects who received at least one dose of study medication. This population was used for the safety analysis. Efficacy analysis was based on a total of 158 subjects; 2 subjects in the ITT population did not have efficacy data collected during treatment (one in the risperidone 1- to 3-mg/day group and one in the risperidone 4- to 6-mg/day group).
Abbreviations: SD = Standard deviation.
The severity of this population's illness is also evident in the mean (SD) baseline CGI-S ratings (placebo, 4.6 [0.7]; risperidone 1–3 mg, 4.7 [0.8]; and risperidone 4–6 mg, 4.5 [0.7]) and in CGAS scores (placebo, 42.2 [12.3]; risperidone 1–3 mg, 39.0 [12.7] and risperidone 4–6 mg, 41.9 [11.6]) (Table 2). CGAS assessments indicated a major impairment at baseline corresponding to an inability to function in one or more of the following areas: Disturbances at home, at school, with peers or in society (i.e., likely to require special schooling and/or hospitalization or withdrawal from school).
Test for no difference between treatments from ANCOVA model with factors treatment and country and screening CGAS score as covariate.
Pairwise comparison: p values and CI associated with the Fisher least significant difference procedure.
Abbreviations: CGAS = Children's Global Assessment Scale; ITT = intent-to-treat; LOCF = last observation carried forward; SD = standard deviation; LS = least squares; SE = standard error; CI = confidence interval.
The 6-week treatment period was completed by 125 (78%) of the subjects with 18 (33%) discontinuing from the placebo-treated group, whereas only 10 (18%) and 7 (14%) discontinued in the risperidone 1- to 3-mg and 4- to 6-mg groups, respectively. The most common reason for discontinuation was insufficient response (24% in the placebo group vs. 5% in the risperidone 1- to 3-mg group and 2% in the risperidone 4- to 6-mg group; Fig. 1). Treatment-emergent AEs caused few discontinuations: Two (4%) subjects in the placebo group, 3 (5%) in the risperidone 1- to 3-mg group, and 4 (8%) in the risperidone 4- to 6-mg group (Fig. 1). The most common treatment-emergent AEs associated with discontinuation were psychosis, dizziness, and somnolence.
The mean (SD) duration of treatment was 36.5 (10.0) days in the placebo group and 38.3 (10.8) and 37.4 (12.1) days in the risperidone 1- to 3-mg and 4- to 6-mg groups, respectively. The median duration of treatment for all three groups was 42 days. Once the fixed target dose was reached, 82% of subjects in the risperidone 1- to 3-mg group had a mode dose of 3 mg/day, 12% had a mode dose of 2 mg/day, and 6% had a mode dose of 1 mg/day. In the risperidone 4- to 6-mg group, 66% of subjects had a mode dose of 6 mg/day, followed by 23% with a mode dose of 5 mg/day and 11% with a mode dose of 4 mg/day.
Primary efficacy
Rapid and highly significant clinical improvement was observed with both risperidone treatment groups, based on change in PANSS total score from baseline versus placebo (p < 0.001 for both comparisons). The between-group difference of the least squares mean change from baseline to end point was −12.0 in favor of the risperidone 1- to 3-mg group and −12.8 in favor of the risperidone 4- to 6-mg group (Fig. 2). Reduction in PANSS scores versus placebo was significant for the 4- to 6-mg risperidone group by day 8 (nominal unadjusted p values ≤0.044) and significant for the 1- to 3-mg group by day 15 (nominal unadjusted p values ≤0.009).
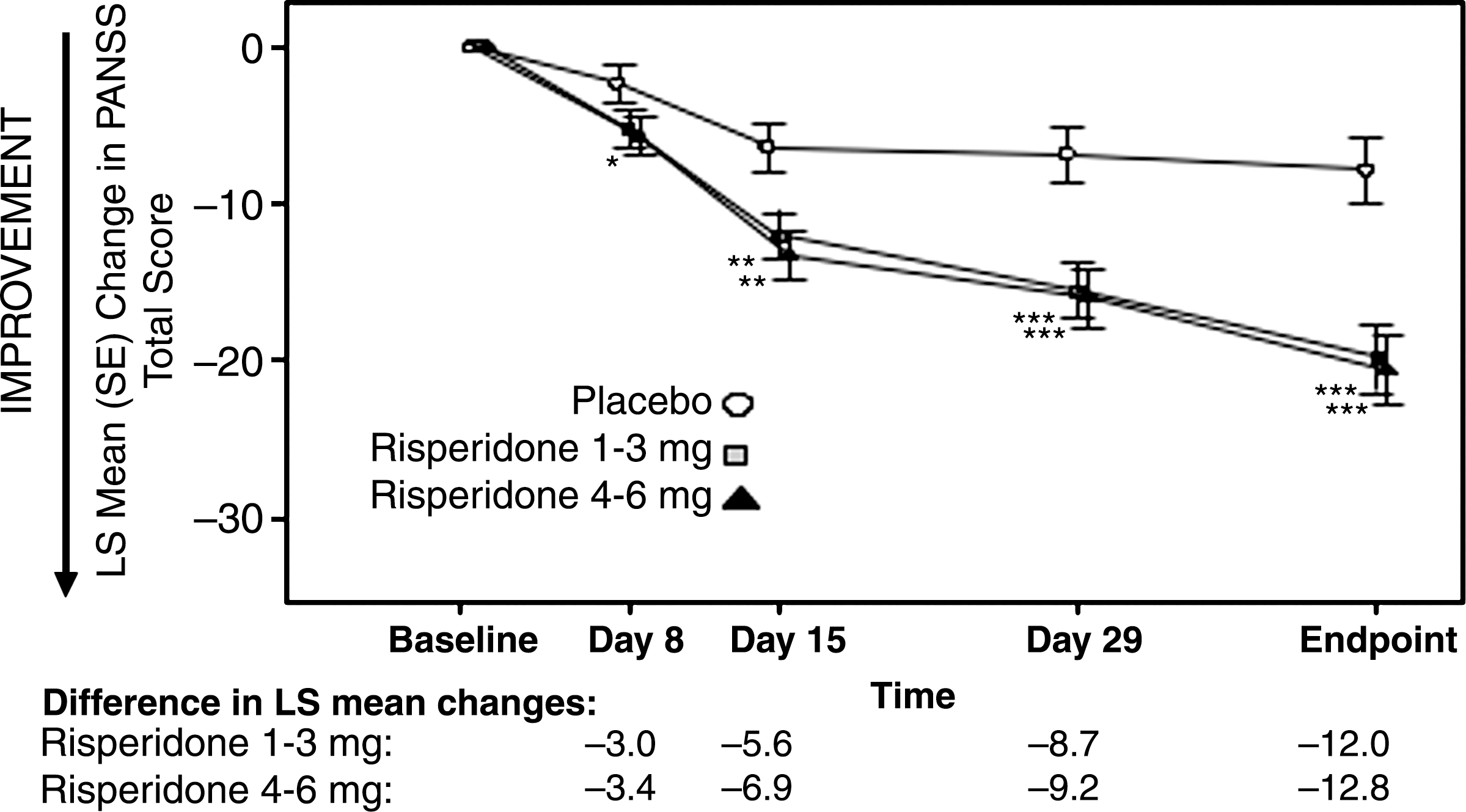
Least-squares mean changes from baseline in PANSS total score over time (ITT subjects with day 43/end-point data [LOCF analysis]). Nominal unadjusted p value (comparison with placebo): (*) p < 0.05; (**) p < 0.01; (***) p < 0.001. A negative mean change from baseline indicates improvement. LS = Least squares; PANSS = Positive and Negative Syndrome Scale; SE = standard error; LOCF = last observation carried forward.
The mixed-effects model revealed a significant treatment effect (p < 0.001) and treatment-by-time interaction effect (p = 0.031). The least-squares estimate of mean change from baseline at day 43 in PANSS total score was −10.3 in subjects who received placebo, −23.0 in subjects who received risperidone 1–3 mg, and −23.7 in subjects who received risperidone 4–6 mg. These results corroborate our LOCF results.
Secondary efficacy
Changes in PANSS factor scores for positive symptoms and negative symptoms at end point are included in Table 3. Mean PANSS factor scores decreased for positive and negative symptoms in both treatment groups when compared with placebo (p ≤ 0.002). Similar findings were observed for PANSS factor scores for disorganized thoughts, uncontrolled hostility/excitement, and anxiety/depression (data not shown).
Test for no difference between treatments from ANCOVA model with factors treatment and country and baseline PANSS score as covariate.
Pairwise comparison: p values and CI associated with the Fisher least significant difference procedure.
Abbreviations: PANSS = Positive and Negative Syndrome Scale; ITT = intent-to-treat; LOCF = last observation carried forward; SD = standard deviation; LS = least squares; SE = standard error; CI = confidence interval.
The clinical response rate (defined as ≥20% reduction from baseline in PANSS total score) was significantly higher at end point in both risperidone treatment groups than in the placebo group: Placebo, 35%; risperidone 1–3 mg, 65% (p < 0.001); risperidone 4–6 mg, 72% (nominal unadjusted p < 0.001) (Fig. 3).
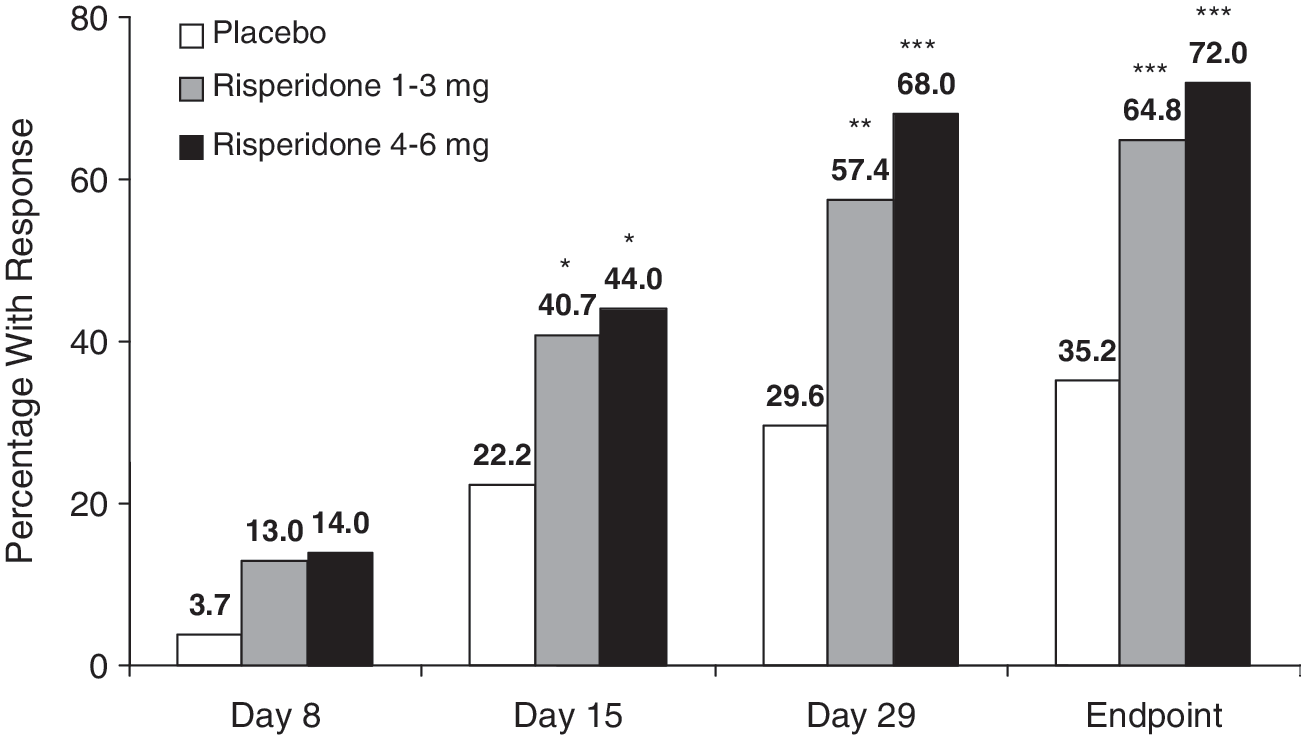
Percentage of subjects with a clinical response at each study time point (ITT subjects with day 43/end point data [LOCF analysis]). Clinical response is ≥20% improvement from baseline in PANSS total score. (*) p < 0.05; (**) p < 0.01; (***) p < 0.001 versus placebo. ITT = Intent to treat; LOCF = last observation carried forward; PANSS = Positive and Negative Syndrome Scale.
CGI-S and CGI-I ratings indicated that subjects who received risperidone 1–3 mg or risperidone 4–6 mg had lower severity of illness at end point and greater improvement relative to subjects who received placebo (Fig. 4A, B). Finally, on measures of global functioning, there were significant improvements in both treatment groups versus placebo (nominal unadjusted p = 0.006 and p < 0.001, respectively) (Table 2). The between-group difference of the least-squares mean changes from screening to end point, compared with placebo, was 7.8 in favor of the risperidone 1- to 3-mg group and 10.8 in favor of the risperidone 4- to 6-mg group.
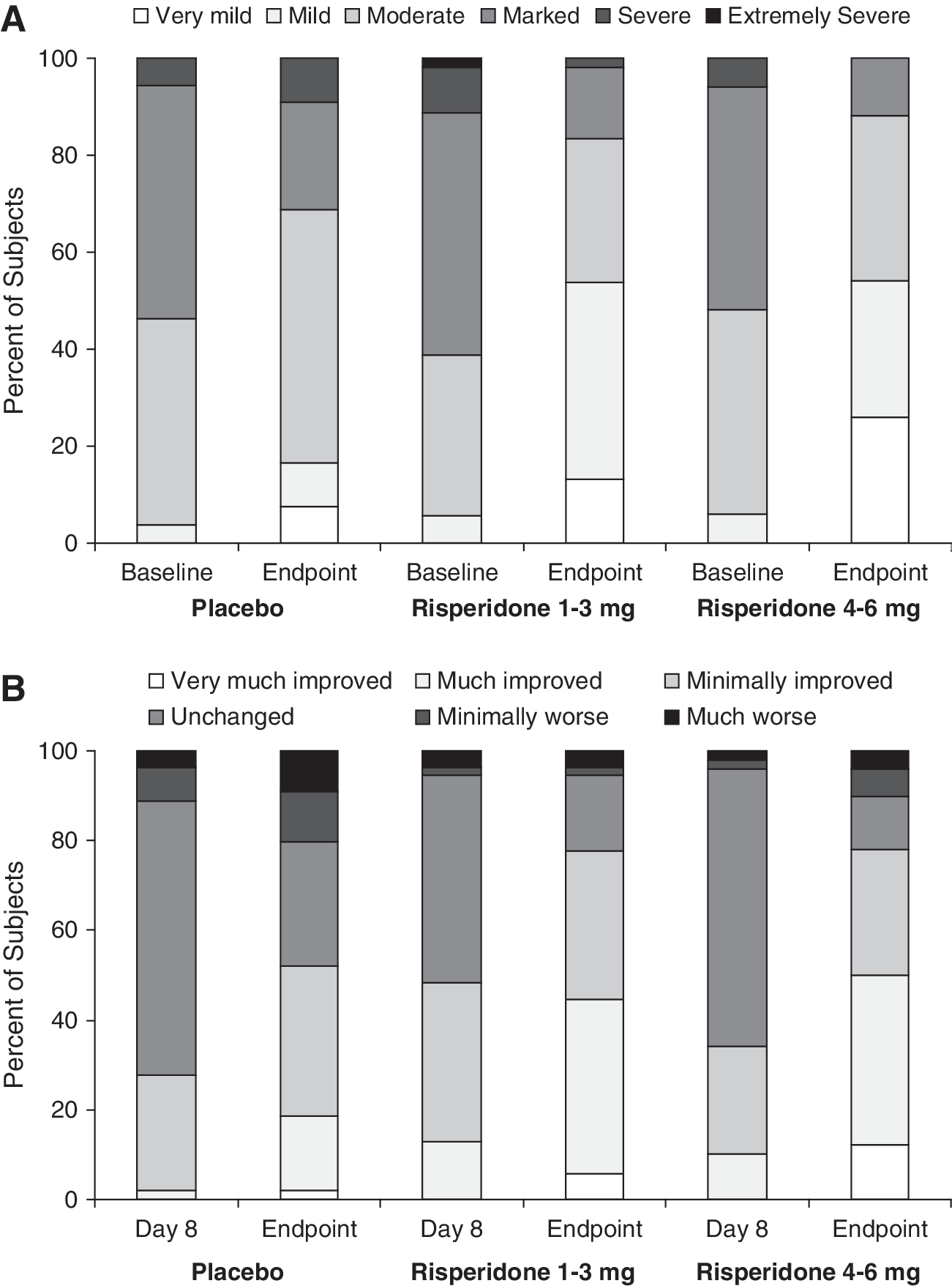
(
Safety
Safety data were available for all 160 subjects who enrolled in the study. Treatment-emergent AEs were reported more frequently in the risperidone treatment groups than placebo (75% and 76% for the 1- to 3-mg and 4- to 6-mg risperidone groups, respectively, vs. 54% in the placebo group) (Table 4). The most common treatment-emergent AEs for the risperidone 1- to 3-mg group were somnolence, agitation, and headache. For the risperidone 4- to 6-mg group, the most common treatment-emergent AEs were extrapyramidal disorder, dizziness, and hypertonia (Table 4). Most AEs were rated as mild to moderate in severity. No suicide-related AEs were reported during the study, and there were no deaths. Serious AEs were experienced by 2 (4%) subjects in the placebo group, 1 (2%) subject in the risperidone 1- to 3-mg group, and 1 (2%) subject in the risperidone 4- to 6-mg group; none were considered related to study medication, and most were related to the underlying psychiatric disorder (e.g., psychosis).
Incidence is based on the number of subjects experiencing at least one AE, not the number of events.
Abbreviations: AE = Adverse events.
EPS-related AEs were summarized as dystonia, parkinsonism, tremor, akathisia, or dyskinesia. Eight (15%) subjects in the placebo group, 18 (33%) subjects in the risperidone 1- to 3-mg group, and 20 (39%) subjects in the risperidone 4- to 6-mg group reported at least one treatment-emergent EPS-related AE. There were no reports of tardive dyskinesia. The severity of EPS at baseline, reflected in AIMS, BARS, and SAS scores, was low; changes from baseline were very small (Table 5).
Higher score indicates worse condition.
A negative mean change from baseline indicates improvement.
Abbreviations: AIMS = Abnormal Involuntary Movement Scale; BARS = Barnes Akathisia Rating Scale; SAS = Simpson–Angus Scale; ITT = intent to treat; SD = standard deviation.
No AEs potentially related to prolactin were reported during the study. At baseline, mean (SD) prolactin levels in male subjects were 21.5 (21.2) μg/L in the placebo group, 21.6 (15.2) μg/L in the risperidone 1- to 3-mg group, and 22.7 (19.9) μg/L in the risperidone 4- to 6-mg group. Corresponding baseline values in female subjects were 24.6 (25.9) μg/L, 39.2 (53.0) μg/L, and 27.9 (28.3) μg/L, respectively. A dose-dependent increase in mean prolactin levels was noted at end point; mean changes were higher in females than in males. Mean (SD) changes from baseline to end point in male subjects were: Placebo group, −3.2 (24.8) μgL; risperidone 1- to 3-mg group, 16.0 (23.7) μg/L; and risperidone 4- to 6-mg group, 26.4 (28.5) μg/L. In female subjects, corresponding mean (SD) changes at end point were −9.2 (24.1) μg/L, 36.9 (41.3) μg/L, and 77.3 (60.8) μg/L, respectively.
Risperidone was associated with nominal differences in absolute weight versus placebo during this 6-week study. Mean weight had increased from baseline to end point by 0.12 kg in the placebo group, 1.3 kg in the risperidone 1- to 3-mg group, and 1.5 kg in the risperidone 4- to 6-mg group. Mean (SD) weight z-scores increased for the risperidone-treated groups, compared with the placebo group: placebo, −0.01 (0.26); risperidone 1–3 mg, 0.09 (0.25); and risperidone 4–6 mg, 0.12 (0.22). Although there was a larger increase in mean weight z-scores in the risperidone 4- to 6-mg group than the risperidone 1- to 3-mg group, these changes were not clinically remarkable. No glucose metabolism-related treatment-emergent AEs were reported. At end point, mean body mass index (BMI) decreased 0.03 kg/m2 in the placebo group, and increased by 0.36 kg/m2 and 0.48 kg/m2 in the risperidone 1- to 3-mg and 4- to 6-mg groups, respectively. No subjects in the study went from <85th BMI percentile at baseline to the ≥95th BMI percentile at end point. There were no clinically meaningful changes in mean electrocardiogram parameters, vital signs, or laboratory values (except prolactin) during the study.
Discussion
This 6-week, double-blind, placebo-controlled study demonstrated that risperidone, at doses of 1–3 mg/day or 4–6 mg/day, was unequivocally superior to placebo in the treatment of adolescents experiencing acute exacerbation of schizophrenia. Improvement in both risperidone dose groups was clinically meaningful, and the changes seen were consistent with those noted in adult schizophrenia subjects treated with risperidone (Borison et al. 1992; Claus et al. 1992; Chouinard et al. 1993; Marder and Meibach 1994; Peuskens 1995). There was no difference in efficacy between the two risperidone treatment groups at end point in the primary and secondary efficacy variables (baseline-to-end point mean change in efficacy scores). Therefore, no additional efficacy benefit was attained by doses higher than 3 mg/day. The earlier onset of benefit from the higher-dose group could be explained by the more rapid titration schedule utilized in this protocol. Once attained, however, improvements were maintained during the remainder of the study for both dose groups.
Schizophrenia is heterogeneous in its clinical presentation, suggesting that many different pathophysiological mechanisms may be contributing to the illness. Evaluation of the PANSS factor scores indicated that both risperidone dose groups achieved significant improvements at end point in all symptom domains (positive symptoms, negative symptoms, disorganized thoughts, uncontrolled hostility/excitement, and anxiety/depression). All other secondary efficacy assessments (clinical response rates, CGI-S, CGI-I, and CGAS) were consistent in demonstrating that both risperidone dose groups provided significant clinical benefit. Within the span of 6 weeks, subjects had a clinically meaningful improvement of their functional performance along with their symptomatic improvement in schizophrenia symptoms.
Overall, risperidone was well tolerated, and there were few discontinuations due to AEs. The overall rate of AEs was similar for the risperidone 1- to 3-mg and 4- to 6-mg dose groups; however, the 4- to 6-mg dose group was associated with a higher incidence of extrapyramidal disorder, dizziness, and hypertonia than the lower-dose group. The qualitative nature of side effects, both reported and measured, was similar to what has been noted in adult schizophrenia subjects treated with risperidone and in other studies of risperidone treatment in children and adolescents with autism, pervasive developmental disorders, or disruptive behavior disorders (Aman et al. 2002; McCracken et al. 2002; Snyder et al. 2002; Reyes et al. 2006a). There were no unexpected findings regarding EPS incidence or severity, prolactin elevations, or weight gain.
Safety results in this 6-week study were generally similar to those of an 8-week randomized study comparing molindone, olanzapine, and risperidone. Although it is difficult to make comparisons because there was no placebo group in that study, risperidone was associated with few reports of EPS, moderate weight gain, and prolactin elevation (Sikich 2008).
Several study limitations must be noted. First, because this was not a fixed-dose study, interpretations regarding the most effective dose must be made cautiously. Although the benefit–risk profile suggests that a dose between 1 and 3 mg/day may be better than a dose between 4 and 6 mg/day, the optimal effective dose was not directly determined. Furthermore, the forced titration to maximally tolerated doses within the two dosage ranges may have resulted in a higher rate of AE reporting than would otherwise have been noted. Finally, although the 6-week trial period was long enough to observe significant improvements with risperidone treatment for psychopathologic symptoms in adolescents with schizophrenia, studies of longer duration are necessary to further assess the continuing efficacy and safety of risperidone treatment on outcomes such as symptom remission and relapse rate. Such studies will allow further monitoring of weight gain, EPS-related AEs, and potential clinical sequelae of prolactin elevation.
Conclusions
In this 6-week study, risperidone treatment was safe and effective at daily doses of 1–3 mg and 4–6 mg in adolescents experiencing acute exacerbation of schizophrenia. The safety and efficacy profile was qualitatively similar to that observed in other studies of risperidone in adult subjects with schizophrenia. The benefit–risk profile suggests that a dose between 1 and 3 mg/day might be optimal for this population.
Footnotes
Disclosures
Dr. Unis worked for Providence Physicians Services at Sacred Heart from 2005 to 2007. He was also a member of the Speakers' Bureau for McNeil, Ortho-McNeil Janssen, and Lilly and was a consultant for Amen Clinic. Currently, Dr. Unis is on the speakers' bureau for Janssen, McNeil, and Lilly and is in contact with Johnson and Johnson, Pfizer, and Wyeth regarding future research trials. Dr. Armenteros was a consultant for Johnson and Johnson at the time of this study. Currently he does not have any financial ties with any pharmaceutical company.
Acknowledgments
The authors wish to recognize Diane Hoffman and Sigrid Malbrain of J&J Pharmaceutical Research and Development for their oversight in conducting the study. We would also like to thank the entire RIS-SCH-302 study team: Leonid Bardenstein, Valeriy Bitenskyy, Vladislav Demchenko, Susan Greer-Day, Elena Grigoryeva, Willis Holloway, Ram Chander Jiloha, Irina Kozlova, Kenneth Lovko, Charles Pinto, Michael Plopper, Mikhail Popov, Yuri Popov, Nadukuru Nooka Raju, G. Prasad Rao, Viktor Samokhvalov, Jitendra Kumar Trivedi, Peter Voloshin, Kashinath G. Yadalam, and Ludmila Yuryeva. The authors also wish to acknowledge the writing and editing assistance provided by Mariana Ovnic, Ph.D., and Helix Medical Communications in the development and submission of this manuscript.
This study was supported by funding from J&J Pharmaceutical Research and Development.