
Abstract
Objectives:
Until the recent approval of methylphenidate (MPH), Japan had no approved treatment for attention-deficit/hyperactivity disorder (ADHD). The need still exists for an effective, safe, nonstimulant treatment. This first placebo-controlled Japan study of an ADHD nonstimulant therapy assessed atomoxetine efficacy and safety to determine the optimal dose for controlling ADHD symptoms in children and adolescents.
Methods:
A total of 245 Japanese children and adolescents, aged 6–17 years and diagnosed with ADHD, were randomly assigned to receive placebo or one of three atomoxetine doses (0.5, 1.2, and 1.8 mg/kg per day) over 8 weeks. Symptoms were assessed with the Japanese Attention-Deficit/Hyperactivity Disorder Rating Scale-IV–Parent Version: Investigator scored and integrated with teacher reports (ADHD RS-IV-J:I/Sch). Adverse events, vital signs, laboratory tests, and electrocardiograms (ECGs) were obtained for safety analysis.
Results:
In all, 234 patients completed the study. Atomoxetine at 1.8 mg/kg per day was significantly superior to placebo in reducing ADHD symptoms (p = 0.01; one-sided). Decreased appetite and vomiting were significantly greater in the atomoxetine treatment groups; however, no clinically significant differences were observed. Two patients discontinued due to affect lability and headache. A linear dose–response and vital signs similar to those from other atomoxetine studies were observed.
Conclusion:
Atomoxetine provides an effective and safe nonstimulant option for the treatment of Japanese pediatric patients with ADHD.
Introduction
ADHD is widely recognized and treated in the United States, where it affects 3–7% of school-aged children (American Psychiatric Association 2000). In Japan, recognition of the core ADHD symptoms has been more limited to physicians working with children who have been referred because of behavioral problems. The concept of ADHD was introduced into Japan in 1994 with the publication of the fourth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-IV), which listed 18 symptoms relevant to a diagnosis of ADHD (American Psychiatric Association 1994). Recently, awareness of the syndrome and its impact on patients and their families are becoming more widely known and have been identified by the Japanese government as high-priority public health issues.
Patients with ADHD in Japan are usually first seen by general practitioners and school nurses, after which they may be referred to pediatricians, psychiatrists, or to one of Japan's regional mental health clinics. The disorder has traditionally been treated in Japan by prescribing stimulants off label. This situation changed in 2007 with the approval of methylphenidate HCl (MPH; Concerta®, Johnson & Johnson), a slow-release formulation for control of the core ADHD symptoms. However, around that time, strict distribution controls on stimulants were introduced due to widespread public concern about abuse potential (Japan Ministry of Health, Labour and Welfare 2007). The potential for stimulant drug abuse remains a key concern for Japan's health-care authorities.
Atomoxetine is a nonstimulant medication for ADHD with negligible potential for drug abuse (Jasinski et al. 2008). The molecule is a highly specific inhibitor of the presynaptic norepinephrine transporter with little affinity for other neurotransmitter transporters or receptors. It has been approved in several countries for the treatment of both pediatric and adult ADHD, including the United States, Canada, most of Europe, Australia, China, Taiwan, and Korea. The efficacy and safety of atomoxetine have also been demonstrated in multiple double-blind, placebo-controlled studies (Michelson et al. 2001; Michelson et al. 2002; Newcorn 2002; Spencer et al. 2002). In each of these studies, atomoxetine was shown to be clinically and statistically superior to placebo in reducing ADHD symptoms in children and adolescents based on information from parents given to investigators in a structured interview. Information from teachers has extended these findings to the school setting and has shown similar outcomes for both efficacy and safety (Weiss et al. 2005). Also, in most trials, the rates of discontinuation due to adverse events were less than 5% (Michelson et al. 2001; Spencer et al. 2001; Biederman et al. 2002; Michelson et al. 2002; Spencer et al. 2002; Buitelaar et al. 2004; Kelsey et al. 2004), indicating that atomoxetine is safe and well tolerated in children and adolescents with ADHD. Prior to this study, no placebo-controlled studies had been conducted in Japanese patients.
Atomoxetine is predominantly metabolized by cytochrome P450 2D6 (CYP2D6) (Ring et al. 2002; Sauer et al. 2003). Most individuals are extensive metabolizers (EM) and possess normal metabolic capacity for CYP2D6 substrates. However, there are some CYP2D6 gene mutations or deletions associated with defective CYP2D6 metabolism, affecting about 7% of the Caucasian population and less than 1% of the Asian population, including the Japanese. These individuals possess two nonfunctional alleles and are classified as poor metabolizers (PM) (Nakamura et al. 1985; Horai et al. 1990; Broly et al. 1991). Although the frequency of PMs is very low in Asian populations, the frequency of intermediate metabolizers (IMs), a subset of EMs with reduced metabolic activity, is relatively high (22%) in Japanese (Iwashima et al. 2007). Alleles for these individuals have decreased enzyme activity (e.g., CYP2D6*10), but metabolic activity is still markedly greater than that seen in PMs. Therefore, there are questions about the relative efficacy and safety of atomoxetine in Japanese patients compared with Western patients.
This study was conducted to meet the urgent need in Japan for a nonstimulant option for the treatment of ADHD. The study plan follows from a specific request by the Japanese health authorities to conduct a trial in Japanese pediatric patients. Overall, the plan was similar to studies conducted in other countries where atomoxetine is already approved. However, a new measure of efficacy was introduced at the request of Japan's health authorities in which the investigator assessed patient improvement based not only on a structured interview with the patient's parent (or primary caregiver) and observation of the patient, but also supplemented by teacher evaluations of classroom behavior. Efficacy in previous studies was based solely on interviews with the parent or primary caregiver (Michelson et al. 2001; Michelson et al. 2002; Newcorn 2002; Spencer et al. 2002), or only on interviews with teachers to confirm outcomes in the school setting (Weiss et al. 2005). The general objectives of this study were to investigate the efficacy and safety of atomoxetine treatment and to determine an optimal dose for Japanese pediatric patients with ADHD.
Methods
Patients
This multicenter study was conducted in 245 Japanese pediatric patients with ADHD at 41 study centers in Japan. Japanese children and adolescents who were at least 6 years old but younger than 18 years of age were eligible to participate if: (1) they met the DSM-IV criteria for ADHD by clinical assessment (American Psychiatric Association 1994) and (2) their diagnosis was confirmed in structured interviews with investigators using the behavior module for ADHD of the Kiddie Schedule for Affective Disorders and Schizophrenia for School- Aged Children–Present and Lifetime Versions (K-SADS-PL) (Kaufman et al. 1997). Also, patients had to have a Clinical Global Impressions–ADHD-Severity (CGI-ADHD-S) assessment score ≥3 (Guy 1976; National Institute of Mental Health 1985) and a symptom severity score at least 1.5 standard deviations (SD) above Japanese pediatric age and gender norms on the Attention-Deficit/Hyperactivity Disorder Rating Scale-IV–Parent Version:Investigator Administered and Scored/Translated and Validated in Japanese (ADHD RS-IV-J:I) (DuPaul et al. 1998; Yamazaki et al. 2001).
Patients were also required to be of normal intelligence (IQ ≥80). For patients younger than 17 years of age, this was assessed by the Wechsler Intelligence Scale for Children–Third Edition (WISC-III). Individual investigators determined normal intelligence in patients 17 years and older.
Important exclusion criteria included patients who took any antipsychotic medication within 26 weeks of study visit 1, had a history of bipolar disorder or psychosis, or were determined by the investigator to be at suicidal risk.
All patients underwent full clinical and laboratory screening; information on patient medical and psychiatric history was collected, and all previous therapy for ADHD was noted. Additionally, blood samples were drawn at the initial study visit and analyzed for the CYP2D6 genotype by testing the *2, *3, *4, *5, *6, *7, *8, and *10 alleles (Genaissance Pharmaceuticals, Morrisville, NC). If patients had two nonfunctional alleles in any combination of *3, *4, *5, *6, *7, or *8 alleles, a PM genotype was assigned; otherwise, an EM genotype was assigned. For the IM subcategory analysis, EMs were divided into four genotypes: (1) ultrarapid metabolizer ([UM] more than two copies of the normal activity allele, e.g., *1/*1XN, *1/*2XN); (2) homozygous EM (either two normal activity alleles, or one normal activity allele and one reduced activity allele, e.g., *1/*2, *1/*10); (3) heterozygous EM (one no activity allele and one normal activity allele, e.g., *1/*5); and (4) IM (either two reduced activity alleles, or one reduced activity allele and one no activity allele, e.g., *5/*10, *10/*10). Two categories were also defined in the IM subcategory analysis to compare the effects of the IM genotype alone (category 1) with those of the IM and heterozygous EM genotypes (category 2).
Written informed consent was received from each patient's parent(s) or legal representative.
Study design and treatments
The study design was based on a previous U.S. study of atomoxetine in children and adolescents with ADHD (Michelson et al. 2001). After an initial 12- to 35-day screening and medication washout period (study period 1), patients were randomized using computer-generated codes into one of three atomoxetine treatment groups (0.5 mg/kg per day, 1.2 mg/kg per day, 1.8 mg/kg per day) or a placebo treatment group for approximately 8 weeks of acute treatment (study period 2). To ensure balance across treatment groups, randomization was stratified by prior use of psychostimulants, age, and ADHD subtype. The study drug was administered as two equally divided doses. To ensure compliance, the drug was taken at meals (before or after), once in the morning, and once in the evening. Patient visits were biweekly during the entire study.
All patients in the atomoxetine arms began treatment at 0.5 mg/kg per day. In the higher dose arms, the drug was titrated with intermediate steps of 0.8 mg/kg per day and 1.2 mg/kg per day at 1-week intervals. Both investigators and patients were blinded to the dose by using capsules that were identical in appearance for all treatment groups. Investigators and patients were also blinded to the patient's CYP2D6 genotype.
The study was reviewed and approved by each site's ethical review board and was conducted in accordance with the ethical standards of the 1975 Declaration of Helsinki as revised in 2000.
Efficacy and safety measures
The primary outcome measure (ADHD RS-IV-J:I/Sch) was designed especially for this study based on a request from Japan's health authorities to include teacher evaluations of patient classroom behavior in the assessment of patient symptoms. ADHD RS-IV is an 18-item scale with each item corresponding to one of the 18 symptoms described in the DSM-IV diagnosis of ADHD (American Psychiatric Association 1994; Du Paul et al. 1998). For the purpose of this study, the scale was translated into Japanese (ADHD RS-IV-J:I) and validated in Japanese patients (Yamazaki et al. 2001). As in the previous U.S. study (Michelson et al. 2001), the scale was scored by the investigator based on a semistructured interview with the patient's parent(s) or primary caregiver. However, in this study the investigator could also integrate scores obtained from teachers (ADHD RS-IV-J:School) of patient classroom behavior during the week prior to the study visit. The ADHD RS-IV-J:I/Sch total score was the investigator assessment of the patient's symptoms based on the parent interview and observation of the patient in conjunction with the completed teacher report (ADHD RS-IV-J:School) that was mailed directly or brought by the parent to the investigator at the scheduled patient visit. Secondary measures of efficacy included the ADHD RS-IV-J:I/Sch Inattentive and Hyperactive/Impulsive subscale scores, and the ADHD RS-IV-J:School total and subscale scores.
Safety and tolerability were assessed by measuring and classifying serious adverse events (SAE), treatment-emergent adverse events (TEAE), treatment adverse drug reactions (ADR; TEAEs judged by investigators to be related to the study drug or study procedure), and patient discontinuations. Changes from baseline to end point in laboratory data, vital signs, and ECG intervals were also used to determine atomoxetine's safety profile. Particularly in the case with ECG QT intervals, various data corrections were used to determine and compare QT intervals (the Bazett, Fridericia, and data driven methods).
Statistical analysis
Entry into this study was planned to continue until approximately 240 patients (60 each for low-dose, mid-dose, and high-dose atomoxetine treatment, and for placebo) were randomized. This sample size provided approximately 90% power for testing the high-dose and placebo groups, and 81% power for testing the mid-dose and placebo groups using Williams test with a one-sided 2.5% significance level (Williams 1971; Williams 1972). All efficacy analyses were performed for patients who met the entry criteria, received at least one dose of study medication, and had baseline and postbaseline measurements (full analysis set). The following data were excluded from the full analysis set: (1) Data obtained after taking a prohibited concomitant medication and (2) data obtained after a major treatment noncompliance.
The primary efficacy measure in this study was the change from baseline to the last observation carried forward (LOCF) end point of the ADHD RS-IV-J:I/Sch total score. For the primary efficacy analysis, the Williams test was performed to compare sequentially the 1.8-, 1.2-, and 0.5-mg/kg per day doses of atomoxetine with placebo using a one-sided 0.025 significance level (Williams 1971; Williams 1972). This is a test for an ordered alternative hypothesis. Specifically, it tests the dose–response relationship and does not inflate the overall probability of a type I error (false positive). Secondary efficacy variables were also assessed using the Williams test.
In the calculation of the ADHD RS-IV-J:I subscales, if more than one item of a subscale was missing, the score for the subscale (and the total score) was considered as missing. If a single item was missing, then the mean score for all other items in the subscale was imputed as the score for the missing item. Linear and quadratic contrasts of least-squares means across dose groups were computed using the analysis of covariance (ANCOVA) model to assess the linear and quadratic relationship for efficacy and safety measures between doses. Treatment differences were also assessed using a mixed model repeated measures (MMRM). The model contained terms for the baseline ADHD RS-IV-J:I/Sch total score, treatment, visit, and treatment-by-visit interaction. The primary contrast of interest from the model was the pair-wise treatment comparisons of the visit 6 scores, which were conducted using contrasts from the repeated measures model. Treatment response was defined as a 25% or greater decrease from baseline in ADHD RS-IV-J:I/Sch total score. Dose–response in binary efficacy and safety measures, such as the percentage of responders or TEAE, were assessed using the Cochran–Armitage Trend test.
The mean and SD were used to describe continuous variables; frequency and percentage were used for categorical variables. A one-sided 0.025 significance level (analysis of Williams test results) and a two-sided 0.05 significance level (all the other analyses) were used. All secondary efficacy analyses were performed for exploratory purposes and no corrections were used for multiple comparisons.
Results
Patients and dosing
Of the 270 patients who entered the study, 245 met the entry criteria and were randomized for treatment into four study arms (Fig. 1). All of these patients were administered at least one dose of the study drug; however, because of prohibited concomitant medication use, an additional 2 patients were excluded from the efficacy analyses, leaving 243 patients in the full analysis set.
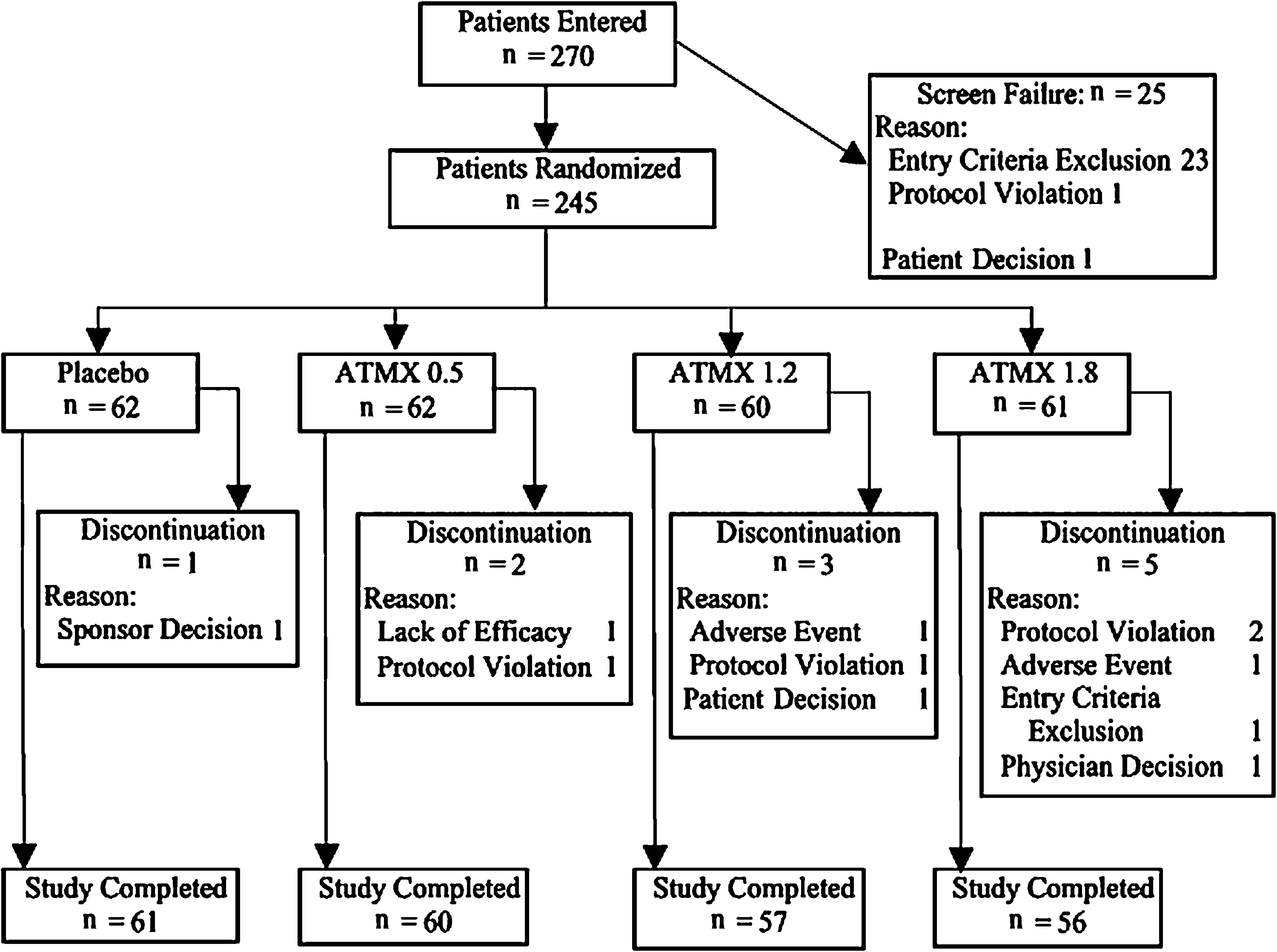
Overview of patient disposition. ATMX = atomoxetine; N = number of patients.
Baseline patient characteristics and symptom severity in all treatment groups were similar and are summarized in Table 1. Mean ages for each treatment group were between 10 and 11 years with males predominant (>80%) in all groups. Inattentiveness was the most prevalent ADHD (61.2% [150/245]), followed by mixed subtype (34.3% [84/245]). Only 4.5% (11/245) of patients were hyperactive/impulsive. Slightly more patients had used stimulants prior to the study (53.9%, 132/245) than those who had not (46.1%, 113/245). The most common co-morbidity was oppositional defiant disorder (13.5%, 33/245); conduct disorder was observed in only 0.8% (2/245) of patients. Other co-morbid diseases were rarely observed, if at all. Two patients were initially classified as EMs with the *2/*5 genotype; however, their plasma atomoxetine concentrations were abnormally high for EMs. Retesting with a detailed genotype test showed that they had the *5/*21 genotype, and they were subsequently categorized as PMs.
Frequencies were analyzed usingthe Fisher exact test.
Means were analyzed using Type III sum of squares analysis of variance.
Category 1: EM (UM, homozygous EM, heterozygous EM), IM, PM.
Category 2: EM' (UM, homozygous EM), IM' (IM, heterozygous EM), PM.
Subjects 401 and 2903 were originally judged as EMs. However, further genetic analysis showed that they were actually PMs.
Abbreviations: (number) = %; ADHD = attention-deficit/hyperactivity disorder; ADHD RS-IV-J:I/Sch = Attention-Deficit/Hyperactivity Disorder Rating Scale-IV–Parent Version:Investigator-Administered and Scored/School (Translated and Validated in Japanese); ATMX =atomoxetine; EM = extensive metabolizer; Hyp/Imp = hyperactive/impulsive; IM = intermediate metabolizer; N = total number of patients; No. = number; PLC = placebo; PM = poor metabolizer; SD = standard deviation.
A more detailed breakdown of patients by CYP2D6 genotype is also shown in Table 1. No statistically significant difference was observed among treatment groups with respect to genotype for either category 1 or 2.
Efficacy
Efficacy outcomes for the primary efficacy measure, ADHD RS-IV-J:I/Sch, are shown in Table 2. At end point, 1.8 mg/kg per day atomoxetine was superior to placebo as seen by the statistically significant change in mean ADHD RS-IV-J:I/Sch total score from baseline (−11.6, p = 0.010; one-sided) compared with placebo. Although statistical significance was not observed at 1.2 mg/kg per day, decreases from baseline scores did show a statistically significant linear dose response (p = 0.008). A repeated measure analysis of ADHD RS-IV-J:I/Sch total scores was also conducted to assess the onset timing of efficacy. After visit 2, a statistically significant reduction (p < 0.05) in mean total score for the atomoxetine 1.8 mg/kg per day treatment group compared with placebo was observed for every visit (visits 3–6). Figure 2 summarizes changes by visit in ADHD RS-IV-J:I/Sch total score for all treatment groups
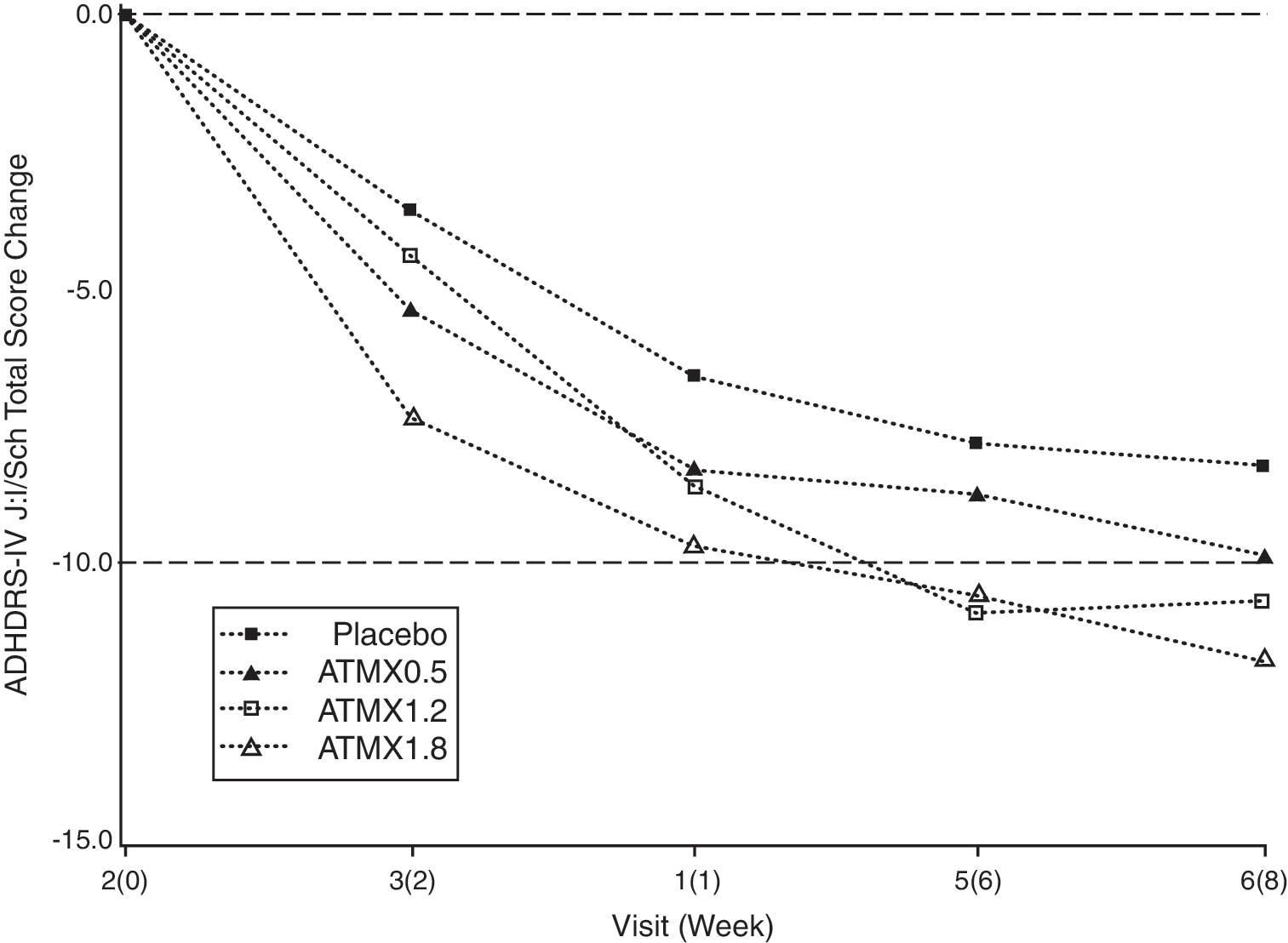
Change in ADHD RS-IV-J:I/Sch total scores over time for all treatment groups. Repeated measure least squares mean (full analysis set). ATMX = atomoxetine; ADHD RS-IV-J:I/Sch = Attention-Deficit/Hyperactivity Disorder Rating Scale-IV–Parent Version: Investigator = Administered and Scored/School (Translated and Validated in Japanese).
ANCOVA mode, change = baseline + treatment; linear contrast, −3 −1 1 3; quadratic contrast, 1 −1 −1 1; n+, Number of patients with both a baseline and at least one post-baseline measurement.
Dose Response p value is derived from the contrasts of the least-square means from ANCOVA model.
p Value (one-sided) is derived from Williams test.
Abbreviations: ADHD RS-IV-J:I/Sch = Attention-Deficit/Hyperactivity Disorder Rating Scale-IV-Parent Version:Investigator-Administered and Scored/School (Translated and Validated in Japanese); LOCF = last observation carried forward; ANCOVA = analysis of covariance; ATMX = atomoxetine; CI = confidence interval; Diff = difference; LCL = lower control limit; n = number of patients; SD = standard deviation; UCL = upper control limit.
Consistent with decreases in ADHD RS-IV-J:I/Sch total score, a statistically significant decrease in the mean score of the secondary outcome measure, ADHD RS-IV-J:I/Sch Inattentive subscale score, was also observed for the atomoxetine 1.8 mg/kg per day treatment group: (−6.8, p = 0.019; one sided). No statistically significant differences were observed in teacher evaluations (ADHD RS-IV-J:School total and subscale scores).
Patients were characterized as responders using definitions specified in the protocol: ADHD RS-IV-J:I/Sch total score decrease of 25% or more from baseline. The percentage of responders in the placebo group was 55.7% (34/61). In the atomoxetine 0.5 mg/kg/day, 1.2 mg/kg/day, and 1.8 mg/kg/day groups, they were 56.5% (35/62), 67.2% (39/58), and 66.7% (40/60), respectively. The percentages of responders for atomoxetine 1.2 mg/kg/day and atomoxetine 1.8 mg/kg/day were very similar.
Safety
Of the 245 patients who entered the study, 183 received at least one dose of atomoxetine. Two SAEs were reported, both in the same patient (8 year-old male; atomoxetine 1.8 mg/kg per day treatment group); these were hospitalization (twice) due to headache and vomiting. However, there were no undue complications and the patient recovered and completed the study without any further problems. Two patients discontinued the study due to adverse events, which were affect lability and headache. Both of these were moderate and the patients recovered without complications.
TEAEs were reported in 78.7% (144/183) of the patients given atomoxetine. The most commonly reported (>10%) TEAEs in the atomoxetine treatment groups were nasopharyngitis (15.8%, 29/183), headache (14.2%, 26/183), and decreased appetite (12.0%, 22/183). Among TEAEs experienced by at least 5% of patients (Table 3), 2 were found to have a statistically significant (p < 0.05) relationship to dose; these were decreased appetite in 12% (22/183) of patients (p <0.001) and vomiting in 8.2% (15/183) of patients (p = 0.022). Other than the single SAE, all TEAEs were either mild or moderate.
All TEAE terms are from MedDRA Version 9.1
Patients who took at least 1 dose of study medication.
p Value is derived from Cochran Armitage Trend test.
Abbreviations: ATMX = atomoxetine; CA Trend Test = Cochran Armitage Trend Test; TEAE = treatment-emergent adverse event; NA = not available.
Treatment ADR were those adverse events judged by the investigator to be related (or possibly related) to either atomoxetine or the study procedure. ADRs were reported in 50.3% (92/183) of patients, and overall showed a statistically significant relationship (p = 0.003) to atomoxetine dose. The most commonly reported ADRs (>10%) were decreased appetite and headache. Only decreased appetite, assessed as an ADR in 11.5% (21/183) of atomoxetine-treated patients, was found to have a statistically significant (p < 0.001) dose–response.
No clinically meaningful differences between treatment groups in laboratory measures were observed. While statistically significant changes were observed in several vital sign measures, none of these was clinically meaningful and there were no related study discontinuations.
Statistically significant (p < 0.05) increases in mean diastolic blood pressure from baseline to end point were observed for all atomoxetine groups compared with placebo. Dose–response was also found to have a statistically significant (p < 0.001) linear component. However, absolute changes were small for all atomoxetine groups (+5.26: 1.8 mg/kg per day; +4.25: 1.2 mg/kg per day; +2.81: 0.5 mg/kg per day) compared with placebo (+0.69) and were not considered to be a cause of concern. Mean systolic blood pressure also showed a statistically significant linear dose–response (p = 0.029); however, no statistically significant difference was found between treatment groups. Although a statistically significant (p ≤ 0.001) increase was also observed in mean heart rate (pulse) for all atomoxetine doses, this was not unexpected given the pharmacologic action of this drug.
Mean height increases in the atomoxetine groups were less than those in the placebo group. Although there was evidence of a statistically significant dose–response (ANCOVA, quadratic contrast; p = 0.036), no statistically significant difference between treatment groups was seen. Statistically significant decreases in mean weight were observed in both the atomoxetine 1.2 mg/kg per day (−0.53 kg, p = 0.002) and 1.8 mg/kg per day (−0.78 kg, p < 0.001) groups compared with placebo (+0.91 kg, p < 0.001). Dose–response was also found to have a statistically significant (p < 0.001) linear component. However, no patient discontinued the study because of weight loss.
There were no serious clinical concerns related to QT interval prolongation. Analysis of electrocardiogram (ECG) results using the data-driven correction for QT interval (QTc) showed mean increases in the QTc interval for all atomoxetine treatment groups; however, all changes were less than 5 msec and none was statistically significant compared with placebo. Similarly, no statistically significant increases in QTc interval were observed with the Fridericia correction, and the only increase (+2.07 msec) was seen in the atomoxetine 0.5 mg/kg per day treatment group. Neither correction gave any evidence of a statistically significant dose–response. Analysis of categorical changes from baseline to end point showed statistically significant trends for increases in QTc interval ≥30 msec with both the Fridericia (p = 0.015) and data-driven (p = 0.034) corrections. However, no cases were reported for a QTc interval >450 msec, nor were any cases reported for a QTc increase from baseline ≥60 msec.
Discussion
The results of this study provide evidence of the efficacy, safety, and tolerability of atomoxetine, a nonstimulant, in Japanese children and adolescents with ADHD. Efficacy increased linearly in a dose-dependent manner with statistically significant superiority of atomoxetine over placebo observed at 1.8 mg/kg per day. Atomoxetine was also tolerated at all treatment doses in this study with no notable safety concerns.
Importantly, this was the first placebo-controlled study in Japan of a nonstimulant ADHD medication. Furthermore, an optimal dose was identified that was safe and well tolerated with statistically significant efficacy. These factors provide strong evidence for the clinical usefulness of atomoxetine and make possible the option of a nonstimulant treatment for both child and adolescent patients with ADHD in Japan, one that lacks the potential for drug abuse.
ADHD RS-IV-J:I/Sch scores showed a statistically significant linear dose–response, reaching an optimal level at 1.8 mg/kg per day. Although statistically significant, efficacy in this study was only 1.8 mg/kg per day. This may be related to two key factors: 1) a comparatively high placebo response and 2) lower patient baseline scores compared with the previous study where 1.2 mg/kg per day was shown to be statistically significant (Michelson et al. 2001). However, the percentage of responders based on a 25% or greater decrease in ADHD RS-IV-J:I/Sch total score and the significant linear dose–response seen in ADHD RS-IV-J:I/Sch scores both point to 1.2 mg/kg per day as a clinically meaningful regimen in Japanese pediatric patients with ADHD.
In another U.S. study (Weiss et al. 2005), information from teachers was used to assess atomoxetine efficacy at once-daily doses up to 1.8 mg/kg per day. This study extended the results of previous studies (Michelson et al. 2001; Michelson et al. 2002; Newcorn 2002; Spencer et al. 2002) in which information from the parent or primary caregiver was the sole basis for assessment of efficacy. The study results confirmed that safety and efficacy outcomes in the school setting were similar to previous study results when reported by teachers rather than parents or primary caregivers. Importantly, the design of the Weiss study was different in that teacher evaluations were determined in structured interviews with the investigators and were the sole basis of efficacy. In the Japan study, completed teacher evaluations (ADHD RS-IV-J:School) were mailed directly or brought by the parent to the investigator at scheduled patient visits. These were supplemental to investigator assessments that were also based on direct information from the parents and observation of the patients.
The safety and tolerability profile of atomoxetine in this study was similar to that seen in studies performed in other countries at doses up to 1.8 mg/kg per day; no major safety concerns and a very low discontinuation rate (Michelson et al. 2001; Weiss et al. 2005; Wang et al. 2007). Specifically, SAEs (two events) were observed in only 1 patient (0.5%, 1/183) and there were just 2 patients discontinuing the study (1.1%, 2/183) due to adverse events. Other than the SAEs seen in this single patient, all AEs were reported as mild or moderate, with no dose–response observed for any of these events. The methodologies used in the double-blind, placebo-controlled studies conducted in the United States, Europe, Australia, and Asia were similar to those used in this Japan study. This lack of major differences between the Japanese ADHD pediatric population and those from other countries indicates that conclusions about the safety and tolerability of atomoxetine from other studies can be applied to Japan. This, combined with data from long-term safety studies, supports extended use in Japanese children and adolescents. A long-term, open-label study of atomoxetine 1.8 mg/kg per day is now under way in Japan to confirm this conclusion.
Limitations
There were some factors in the study design that limit the conclusions that can be drawn from these results. Because this was the first placebo-controlled study in Japan of a nonstimulant compound for treatment of ADHD in pediatric patients, there were no prior results in Japanese children to gauge the most appropriate selection of patients, titration schedule, or assessment of ADHD symptom improvement. All of these had to be based on the results from similar studies conducted in non-Japanese populations.
The primary measure of efficacy in this study was the change from baseline in ADHD RS-IV-J:I/Sch total score. Unlike studies conducted in other countries, where the investigator assessed ADHD symptoms based solely on information from the parent or primary caregiver, investigators in the Japan study determined this score by integrating information from both parents and teachers. This was the first time such an integrative system was used. However, teacher evaluations (ADHD RS-IV-J:School total score) were completed independently by the teacher, without any direct contact with the investigator. This was fundamentally different from the information obtained from parents that was based on structured interviews with the investigators. Furthermore, in this study inattention was the most prevalent subtype (61.2% of patients) and may have made it more difficult for teachers to assess patient improvement compared with the hyperactive/impulsive or mixed subtypes. This could have contributed to the lack of consistency in teacher evaluations and adversely affected investigator assessments of ADHD RS-IV-J:I/Sch total score.
Another issue was the high placebo response seen in this study. This was probably due to differences between teacher and investigator assessment scores of ADHD symptom improvement in the placebo group. One post hoc subgroup analysis showed that there were 37 patients where teacher/investigator assessments differed by 9 points or more at baseline (−10.1 ± 7.3 mean change from baseline) and 24 patients where assessments differed by less than 9 points at baseline (−5.0 ± 5.6 mean change from baseline). This inconsistency might have been due to teachers interpreting inattention as more subdued behavior in the classroom, and thus as an improvement, which would have contributed to the high placebo response rate. The actual root cause for the high placebo rate is unknown, but this difference between teacher and investigator assessment of ADHD RS-IV-J scores was most likely a main factor in the phenomenon.
Finally, there was the low baseline ADHD RS-IV-J score seen in this study compared with the pivotal U.S. study (Michelson et al. 2001). This was probably caused by the large proportion of inattentive subtype patients (61.2%) compared with only 4.5% hyperactive/impulsive subtype and 34.3% mixed subtype patients. Although this does not represent the actual prevalence of the inattentive subtype in Japan overall, it would have made significance harder to achieve at 1.2 mg/kg per day, and is most likely the main reason why statistically significant efficacy was not observed at this dose level.
Clinical Implications
This study can be seen as the first step in providing Japanese physicians with an efficacious, safe and tolerable nonstimulant option in the treatment of Japanese ADHD pediatric patients. In this first study, statistically significant efficacy was observed at 1.8 mg/kg per day. However, this was necessarily a short-term placebo-controlled study (8 weeks treatment) performed to demonstrate efficacy and the absence of any immediate concerns related to safety and tolerability. Further testing at this dose level is needed to confirm safety and tolerability over the longer administration time frames expected in the treatment of ADHD. We are now conducting a long-term, open-label study at 1.8 mg/kg per day to confirm our initial conclusions on safety and efficacy.
Conclusions
The results from this study, the first placebo-controlled trial in Japan of a nonstimulant medication to control core ADHD symptoms, demonstrated that atomoxetine at a fixed dose of 1.8 mg/kg per day can significantly reduce core symptoms in Japanese pediatric patients with ADHD. Although the duration of atomoxetine treatment in this first study was set at 8 weeks, our findings were comparable to results from similar studies worldwide (Michelson et al. 2001; Spencer et al. 2001; Biederman et al. 2002; Michelson et al. 2002; Spencer et al. 2002; Buitelaar et al. 2004; Kelsey et al. 2004), atomoxetine was found to be safe and well tolerated up to 1.8 mg/kg per day.
Footnotes
Disclosures
Michihiro Takahashi is an employee of Eli Lilly Japan K.K.; he is Lead Clinical Research Physician for Neuroscience Products. Yasushi Takita is an employee of Eli Lilly Japan K.K.; he is a statistician working on the Project Statisticians Team. Kosuke Yamazaki was funded for his work on this study by Eli Lilly Japan K.K. Takashi Hayashi has had funding support from Eli Lilly Japan K.K. and Janssen Pharmaceutical K.K. (Japan); funding for his work on this study was provided by Eli Lilly Japan K.K. Hironobu Ichikawa was funded for his work on this study by Eli Lilly Japan K.K. directly to his hospital, the Tokyo Metropolitan Umegaoka Hospital (Tokyo, Japan). Yasuko Kambayashi was funded for her work on this study by Eli Lilly Japan K.K. Tatsuya Koeda has had funding support from Eli Lilly Japan K.K. and Janssen Pharmaceutical K.K. (Japan); funding for his work on this study was provided by Eli Lilly Japan K.K. Junichi Oki was funded for his work on this study by Eli Lilly Japan K.K. Kazuhiko Saito has had funding support from Eli Lilly Japan K.K., Janssen Pharmaceutical K.K. (Japan), Astellas Pharma, Inc. (Japan), and Meiji Seika, Ltd. (Japan); funding for his work on this study was provided by Eli Lilly Japan K.K. Kenzo Takeshita was funded for his work on this study by Eli Lilly Japan K.K. Albert J. Allen works as a Medical Director at Lilly Research Laboratories, Indianapolis, Indiana.
Acknowledgments
The authors acknowledge Nobuya Matsuhara (Eli Lilly Japan) for assistance in confirming data integrity and Gregory H. Smith (consultant) for editorial assistance with the manuscript.
This research was funded by Eli Lilly Japan K.K.