
Abstract
Objective:
The aim of this study was to assess the effectiveness and safety of lisdexamfetamine dimesylate (LDX) in children with attention-deficit/hyperactivity disorder (ADHD).
Method:
This was a 7-week, open-label study evaluating 20, 30, 40, 50, 60, or 70 mg/day LDX in 318 children aged 6–12 years with ADHD. The ADHD Rating Scale IV (ADHD-RS-IV) was the primary efficacy assessment. Secondary measures included the Clinical Global Impressions–Improvement (CGI-I), Expression and Emotion Scale for Children (EESC), and Behavior Rating Inventory of Executive Function (BRIEF). Safety assessments included treatment-emergent adverse events (TEAEs), vital signs, and electrocardiograms.
Results:
At end point, mean (standard deviation [SD]) improvement from baseline in ADHD-RS-IV total score was 28.6 (10.9) (p < 0.0001). Most subjects (89.9%) were rated “improved” (i.e., CGI-I 1 or 2). Improvements from baseline were observed in the EESC total and subscale scores (p ≤ 0.0002). LDX treatment resulted in significant improvement on the Global Executive Composite, Behavioral Regulation, and Metacognition indices of the BRIEF (p < 0.0001). TEAEs (incidences ≥10%) were decreased appetite, decreased weight, irritability, insomnia, headache, upper abdominal pain, and initial insomnia.
Conclusions:
LDX was effective and generally well tolerated with a safety profile consistent with long-acting stimulant use. There was overall improvement in ADHD symptoms and executive function measures and no worsening of emotional expression measures.
Trial Registration:
clinicaltrials.gov Identifier: NCT00500071.
Introduction
Executive function represents a collection of cognitive processes that affect goal-directed behavior, play a role in self-regulation, and control emotional functioning (Barkley 1997; Gioia et al. 2000; Gioia et al. 2002). Executive function includes inhibition, set-shifting, regulation of behavior, organizing, planning, working memory, and emotional regulation (Pennington et al. 1996; Barkley 1997; Mares et al. 2007). Impairments in executive functioning have been posited to play a role in the symptomatology of ADHD (Pennington et al. 1996; Barkley 1997; Swanson 2003; Biederman et al. 2006; Brown 2006). Furthermore, psychometrically defined deficits in executive functioning have been associated with poor outcomes in patients with ADHD (Biederman et al. 2004; Biederman et al. 2006). Aspects of executive functioning can be assessed via neuropsychological tests in a laboratory setting. For instance, the Stroop test can be used to assess set shifting, while the Tower of London task can be used to evaluate planning ability (Pennington et al. 1996; Swanson 2003). The Behavior Rating Inventory of Executive Function (BRIEF), however, was developed to assess real-world executive function behaviors in the home and/or school as assessed by parents and teachers using a behavior rating scale rather than relying on clinic-based performance tests (Gioia et al. 2002). A rating scale that assesses executive function behaviors as perceived by parents may capture deficits that are not evident on isolated neuropsychological tests. Prior studies evaluating laboratory measures of executive function have demonstrated considerable variability such that the finding of deficits on neuropsychological tests is generally predictive of ADHD, but negative findings (e.g., scores in the normal range) do not rule out the condition. Thus, many patients with ADHD fall within the normal range on single measures and some test normally on multiple measures (Doyle et al. 2000; Doyle 2006). The BRIEF has been successfully used in studies of children with ADHD (Jarratt et al. 2005; Mares et al. 2007; McCandless et al. 2007; Qian et al. 2007b), but, to our knowledge, it has not been widely used in prospective trials to assess the effects of medications used to treat ADHD in children (Qian et al. 2007a).
Alterations in emotional regulation secondary to difficulties in behavioral inhibition have been hypothesized to play an integral role in the executive function deficits associated with ADHD (Barkley 1997). Others posit that managing frustration and regulating emotion is one of several cognitive functions impacted by deficits in executive functioning (Brown 2006). Children with ADHD may exhibit temper outbursts, mood lability, and dysphoria (American Psychiatric Association 2000). Using data from the 2003 National Health Interview Survey, Strine et al. demonstrated that approximately one quarter of children with ADHD were reported to exhibit high levels of emotional problems (Strine et al. 2006). This was more than 3.5 times greater than the prevalence in children without a history of ADHD. Given that emotional regulation and modulation of frustration have been recognized as core executive function behaviors, a scale for “emotional control” is included among the eight BRIEF scales that assesses a child's ability to manage emotional responses (Gioia et al. 2000; Gioia et al. 2002; Mares et al. 2007; Sullivan et al. 2007).
Stimulants are considered first-line pharmacotherapy for pediatric patients with ADHD, and their efficacy is well recognized (American Academy of Pediatrics Subcommittee on Attention-Deficit/Hyperactivity Disorder and Committee on Quality Improvement 2001; Greenhill et al. 2002; Brown et al. 2005; Pliszka et al. 2006; Spencer et al. 2006). However, stimulants' effects on emotional expression and executive function have not been well documented (Kempton et al. 1999; Fallu et al. 2006; Kratochvil et al. 2007). A reported side effect of stimulants is the restricting or blunting of emotional expression (e.g., the outward appearance of a child's affect and expressed emotions) (American Academy of Pediatrics Subcommittee on Attention-Deficit/Hyperactivity Disorder and Committee on Quality Improvement 2001; Perwien et al. 2008). Children that have been prescribed stimulants have sometimes been described as “zombie-like,” and this effect may potentially impact parental decisions regarding treatment regimens for ADHD (Greenhill et al. 2002; Kratochvil et al. 2007). The impact of stimulants on emotional expression may be difficult to separate from the emotional effects seen when medication wears off (“rebound” phenomena). There are reports from both parents and clinicians of intense late afternoon rebound effects as the drug wears off (Greenhill et al. 2002). In some patients, short-acting stimulants have been described as being associated with sadness, crying, and irritability when the last daily dose of medication has worn off (Carlson et al. 2003). The potential for fluctuations in symptom amelioration and the short duration of effect seen with immediate-release stimulants have prompted the development of long-acting stimulants (Connor et al. 2004).
Emotional expression encompasses both positive and negative aspects (Perwien et al. 2008). Positive aspects of emotional expression include happiness, creativity, and confidence while negative aspects include dysphoria and flat moods. The Expression and Emotion Scale for Children (EESC) was designed to evaluate both the negative and positive aspects of emotional expression in children when taking medication (Perwien et al. 2008). In a cross-sectional study, no differences in emotional expression as assessed by the EESC were seen in children with ADHD receiving atomoxetine compared to those taking stimulants (Kratochvil et al. 2007). However, to our knowledge, there have been no prospective studies that have evaluated the impact of stimulants on emotional expression in children with ADHD as assessed by the EESC.
Changes in emotional expression and executive functioning can impact quality of life and, as such, these domains should be considered when selecting treatment regimens for children with ADHD (Biederman et al. 2006; Perwien et al. 2008). Currently, there is a paucity of data prospectively examining the impact of various drugs used to treat ADHD on these domains.
Lisdexamfetamine dimesylate (LDX) is a long-acting prodrug stimulant indicated for the treatment of ADHD in children and in adults. LDX is a therapeutically inactive molecule converted to l-lysine and therapeutically active d-amphetamine after oral administration (Biederman et al. 2007a). In a crossover analog classroom study in children with ADHD, LDX treatment resulted in significant reductions in symptoms of ADHD as assessed by the Clinical Global Impressions (CGI) scale, Swanson, Kotkin, Agler, M-Flynn, and Pelham (SKAMP) Rating Scale, and Permanent Product Measure of Performance (PERMP) scores compared with placebo (Biederman et al. 2007a). Treatment effects were observed at 12 hours postdose, the last time point measured. In a large, multicenter, randomized, placebo-controlled trial, efficacy was demonstrated by clinician measures including the ADHD Rating Scale Version IV (ADHD-RS-IV) (Biederman et al. 2007b), as well as efficacy versus placebo throughout the day, up to 6 p.m., as measured by parent ratings on the Conners' Parent Rating Scale–Revised: Short Form (CPRS-R). In a second laboratory school study, LDX demonstrated efficacy up to 13 hours postdose (Wigal et al. 2009).
A recent long-term study demonstrated that LDX was safe and effective in reducing ADHD symptoms in school-aged children for up to 1 year (Findling et al. 2008). In short-term trials of LDX in children with ADHD, LDX was also generally well tolerated and displayed a safety profile consistent with long-acting stimulant use (Biederman et al. 2007a; Biederman et al. 2007b; Findling et al. 2008). In the large, multicenter double-blind, placebo-controlled study, the most common treatment-emergent adverse effects (TEAEs) seen with LDX treatment (i.e., those with a >10% incidence in LDX treatment groups) were decreased appetite, insomnia, upper abdominal pain, and headache (Biederman et al. 2007b).
Prior clinical trials of LDX in children, including two trials using a forced-dose titration design and two trials utilizing a dose-optimization design, have evaluated the efficacy and tolerability of LDX in dosages of 30, 50, and 70 mg/day (Biederman et al. 2007a; Biederman et al. 2007b; Findling et al. 2008; Wigal et al. 2009). These dosages of LDX are converted to 8.9 mg, 14.8 mg, and 20.8 mg of d-amphetamine, respectively. Recently approved dosages of 20, 40, and 60 mg/day of LDX (converted to 5.9 mg, 11.9 mg, and 17.8 mg of d-amphetamine, respectively) may allow for greater dosing flexibility and may facilitate titration to achieve maximum efficacy and tolerability while minimizing AEs in patients with ADHD. This is the first study to evaluate the effectiveness, safety, and tolerability of the expanded range of dosage strengths of LDX in children with ADHD dosed to optimal effect. This study also prospectively evaluated the effects of LDX on executive function and emotional expression using the BRIEF and EESC scales, respectively.
Methods
This prospective, open-label, multicenter, dose-optimization, and maintenance study enrolled subjects from 42 sites in the United States between June, 2007, and January, 2008. The purpose of this study was to evaluate the safety, tolerability, and effectiveness of LDX with an expanded dose range during dose optimization of treatment in children with ADHD. As such, this study would serve as a heuristic reflection of the prescribing practices of most clinicians. An additional aspect of this study is the evaluation of response to LDX on measures of executive function and emotional expression.
Participants
The study enrolled children aged 6–12 years with a primary diagnosis of ADHD by DSM-IV-TR criteria. Subjects were required to have at least moderate symptoms of ADHD as assessed by a baseline ADHD-RS-IV total score of at least 28. Other inclusion criteria included age-appropriate intellectual functioning and blood pressure (BP) measurements within the 95th percentile for age, gender, and height. An exclusion criterion included a co-morbid psychiatric diagnosis with significant symptoms that would, based on the evaluation of the investigator, be a contraindication for LDX or would confound assessments of efficacy and safety. Additional exclusion criteria included conduct disorder; history of hypersensitivity, allergy, intolerable response, or failure to respond to amphetamine therapy; weight less than 50 pounds; and body mass index greater than the 98th percentile. Other exclusion criteria included a history of seizures or tic disorder, a clinically significant electrocardiogram (ECG) abnormality, known structural cardiac abnormality, concomitant medications that affect the central nervous system (excluding ADHD medications, which were discontinued prior to receiving study-related treatment), and being effectively treated on current ADHD medication with acceptable tolerability.
Written informed consent was obtained from each patient's legal guardian, and assent was obtained from each child prior to study-related procedures being performed. The study protocol was approved by the institutional review board at each study center, and the study was performed in accordance with the International Conference on Harmonisation (ICH) of Good Clinical Practice (GCP) principles of the World Medical Association's Declaration of Helsinki.
Study design and visits
The present study had three phases: A screening and washout period lasting approximately 2 weeks, an open-label treatment phase lasting approximately 7 weeks, and a 30-day follow-up period. Following the baseline visit of the open-label treatment phase (visit 0), all subjects were to begin treatment with once-daily LDX at a dosage of 20 mg. Subjects subsequently reported to the study site weekly for 7 weeks (i.e., visits 1 through 7). At each visit, patients were evaluated and their response to treatment was assessed. Patients demonstrating an ineffective response, defined as not having achieved at least a 30% reduction in ADHD-RS-IV total score from baseline and a CGI–Improvement (CGI-I) score of 3 or greater, had their dose escalated in 10-mg weekly increments each week up to 70 mg/day if tolerability was acceptable. No dose escalation was permitted beyond week 5. Patients experiencing intolerable side effects were downtitrated to the next available dose. Only one downward dose taper was permitted, and subjects with intolerable side effects at a lowered dose were discontinued from the study. Subjects were deemed to have an acceptable response if they achieved at least a minimum 30% reduction in ADHD-RS-IV total score from baseline and a CGI-I score of 1 or 2 with tolerable side effects. These subjects had their dose of LDX maintained for the rest of the study. The protocol of this dose-optimization study also allowed dosages to be increased (up to week 5) in 10-mg increments if, in the clinician's judgment, a subject could potentially receive additional symptom reduction beyond an “acceptable” response. All subjects were to complete an end-of-study/early termination visit at week 7 (or earlier if discontinued from the study).
Assessment measures
The primary efficacy assessment of this trial was the ADHD-RS-IV. The clinician-completed ADHD-RS-IV contains 18 items reflective of DSM-IV-TR ADHD criteria. The items comprise a total score that includes two subscales: Inattention and hyperactivity/impulsivity. Each item is scored between 0 (never or rarely) and 3 (very often). The ADHD-RS-IV was assessed at baseline and all weekly study visits thereafter.
Secondary efficacy assessments included the CGI, Parent Global Assessment (PGA), BRIEF, and EESC. The CGI was assessed weekly, with clinicians completing the CGI–Severity (CGI-S) at baseline and the CGI-I thereafter. The clinician-rated CGI-S allowed for a global evaluation of a subject's severity of illness at baseline, using a 7-point scale ranging from 1 (normal, not at all ill) to 7 (among the most extremely ill subjects). The CGI-I was used to assess patient improvement, also using a 7-point scale (1 [very much improved] to 7 [very much worse]). For the purpose of analysis, CGI-I scores were dichotomized into two categories: Improved (1 [very much improved] and 2 [much improved]) and not improved (all other categories). The PGA is a parent-rated instrument designed to assess parental impressions of their child's improvement from baseline. At baseline, parents provided notes regarding their child's pretreatment behavior. Thereafter, parents rated their child's improvement on a 7-point scale from 1 (very much improved) to 7 (very much worse) weekly. Scores were dichotomized for analysis in a manner identical to that for the CGI-I.
The EESC, a validated measure of emotional expression, was completed at baseline and at the end of study or early termination visit (Kratochvil et al. 2007; Perwien et al. 2008). Each item of the 29-item EESC is rated on a 5-point scale from 1 (not true at all) to 5 (very much true). The EESC total score ranges from 29 to 145, with higher scores reflecting poorer emotional outcomes. The EESC contains three subscales: Emotional Flatness (EF), Positive Emotions (PE), and Emotional Lability (EL). As with the EESC total score, higher scores on these subscales reflect poorer emotional outcomes. The EF subscale asks respondents to answer 10 questions that assess, for example, if the patient “seems down” or “is sluggish,” and scores range from 10 to 50 (Perwien et al. 2008). The PE subscale contains 13 questions about topics such as “friendliness” and “enthusiasm” with scores that can range from 13 to 65, whereas the EL subscale assesses 5 items including “mood swings” and “crankiness,” with scores ranging from 5 to 25 (Perwien et al. 2008). One item regarding “range of emotions” is not included in any of the subscales but is part of the total composite score. It should be noted that the EESC has been recently revised such that the instrument used in the present study should be referred to as Version 1 (Kratochvil et al. 2007; Perwien et al. 2008). This earlier version of the instrument has been used in a cross-sectional study to investigate the effects of ADHD medications and in a prospective study of atomoxetine in children aged 6–12 years (Kratochvil et al. 2007).
The BRIEF is a validated instrument that measures components of executive functioning in children 5–18 years of age (Gioia et al. 2002; McCandless et al. 2007). This parent-rated measure contains 86 items, which parents score as 1 (never), 2 (sometimes), or 3 (often), and was assessed at baseline and at the last study visit (Gioia et al. 2002). Although several factor models for the BRIEF have been proposed (Gioia et al. 2002), the present study used the model originally developed for the scale. The measure is made up of 8 scales, which in turn comprise 2 indices: A Behavioral Regulation Index (BRI) (inhibit, shift, and emotional control) and a Metacognition Index (MI) (initiate, working memory, plan/organize, organization of materials, and monitor) (Gioia et al. 2000). The two indices comprise a Global Executive Composite (GEC) score. Raw scale scores are transformed into T-scores for interpretation such that a score of 50 represents the mean for the normative group and 10 points represents 1 standard deviation (SD) from the mean (Sullivan et al. 2007). Higher scores represent greater levels of dysfunction and scores of 65 or greater are considered to be potentially clinically significant (Gioia et al. 2000). The BRIEF has previously been used to assess deficits in executive functioning exhibited by children with ADHD (Mares et al. 2007; McCandless et al. 2007; Sullivan et al. 2007).
The Medication Satisfaction Questionnaire (MSQ), a nonvalidated measure, was completed by parents at the final study visit. Parents were asked three questions regarding their satisfaction with LDX namely: (1) Their level of satisfaction with LDX treatment, (2) a comparison of LDX with their child's previous ADHD drug treatment, and (3) their likelihood of continuing to use LDX for their child.
Safety assessments in the present study included the recording of reported treatment-emergent adverse events (TEAEs) at every visit. Patients' vital signs and weight were assessed at all study visits, while height was assessed at screening and the last study visit. ECGs were collected at screening, baseline, and the end of study visit, and parameters assessed included heart rate as well as PR, QRS, and QT intervals. Additionally, QT intervals were corrected for heart rate using the Bazett formula (QTcB) and Fridericia formula (QTcF).
Data analysis
A sample size of approximately 300 patients was planned for the present study based on the sample size of a prior pivotal study (Biederman et al. 2007b), but no formal power analysis was performed. As such, the present study was not prospectively powered nor designed to provide dose–response comparisons. Effectiveness was assessed in the intention-to-treat (ITT) population, defined as all subjects who received at least one dose of LDX and had at least one follow-up ADHD-RS-IV assessment. Safety was assessed in the safety population, defined as all subjects who took at least one dose of LDX.
The primary efficacy variable was the change in ADHD-RS-IV total score from baseline to end point, which was analyzed by a one-sample t-test. End point was defined as day 49, or, for subjects that discontinued early, the last valid ADHD-RS-IV score after baseline (last observation carried forward). Secondary efficacy analyses using one-sample t-tests were performed on ADHD-RS-IV total score and subscale changes from baseline scores of those subjects completing each weekly assessment. These analyses were also performed separately by gender. Other secondary efficacy measures (i.e., CGI-S, CGI-I, PGA, and MSQ) were summarized using descriptive statistics. Post hoc analyses using the one-sample t-test were conducted on EESC and BRIEF total and subscale score change from baseline to end of study for all LDX dose levels combined.
For safety analyses, AEs were coded using the Medical Dictionary for Regulatory Affairs (MedDRA), Version 10 (Northrop Grumman Corporation). AEs were classified as TEAEs if they started on or after the first day of treatment with LDX and no more than 3 days after the last date of treatment. The change from baseline for vital sign measurements, height, and weight was summarized using descriptive statistics. A priori outlier criteria were established for vital signs and included: Systolic blood pressure (SBP) <90 mmHg or >120 mmHg; diastolic blood pressure (DBP) <50 mmHg or >80 mmHg; and pulse ≤45 bpm or ≥130 bpm. ECG parameters were also summarized by descriptive statistics as well as by outlier criteria including PR interval ≥180 msec, QRS interval ≥100 msec, QT interval ≥450 msec, QTcB ≥450 msec, QTcF ≥450 msec, change in QTcB or QTcF ≥30 msec, and heart rate ≤45 bpm or ≥130 bpm. Distribution of changes in vital signs, ECG parameters, and weight were assessed in post hoc analyses by descriptive statistics, including means, medians, 25th and 75th percentiles, interquartile range (defined as the range of values between the first and third quartile), and extreme values. Compliance was assessed, a priori (as part of the prospective statistical plan), as percent compliance by counting number of capsules taken divided by the number of days of dosing.
Results
Subject demographics and disposition
A total of 318 children were enrolled in the present study. The safety population was composed of 317 children secondary to the discontinuation of 1 subject as a result of a protocol violation (i.e., failed to return and lost to follow-up after initial dispensing of drug). The ITT population was composed of 316 participants, and 278 subjects completed the study. Forty subjects discontinued before study completion: 9, 4, 1, 4, 3, 5, 9, and 5 subjects in weeks 1 through 8, respectively. The most common reasons for discontinuation were loss to follow-up (n = 17 [5.3%]) and AEs (n = 13 [4.1%]) (Fig. 1). In the safety population, 224 subjects (70.7%) were male and 93 subjects (29.3%) were female. Baseline characteristics between the two genders were similar (Table 1).
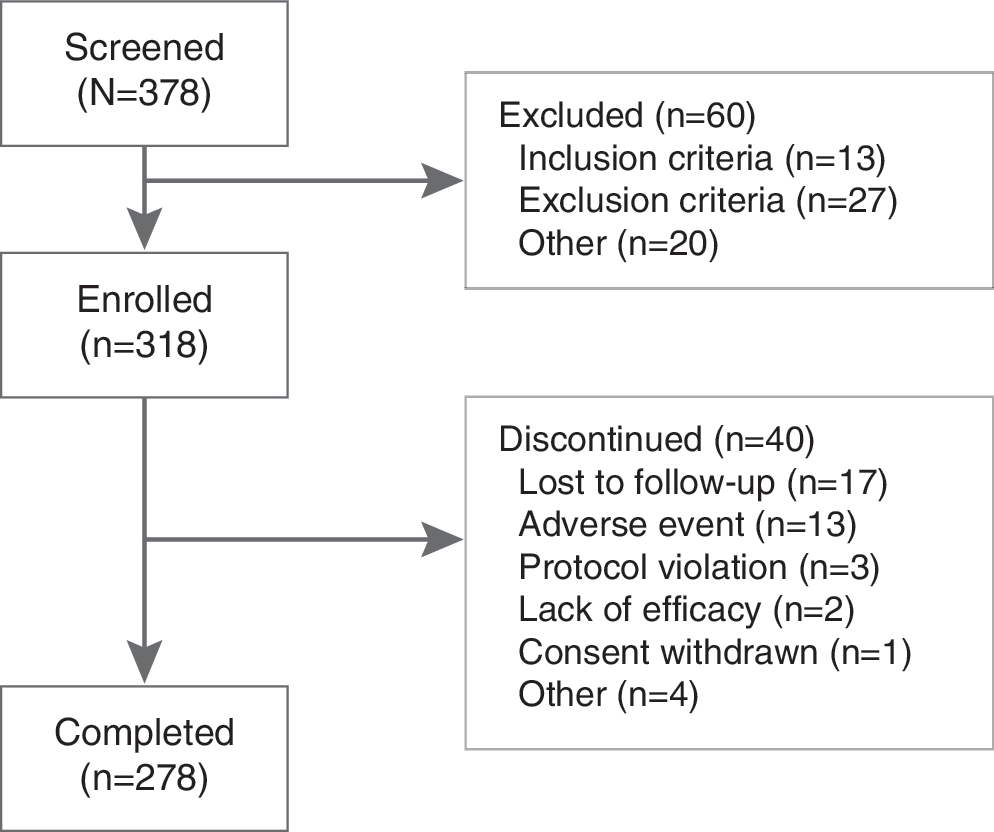
Subject disposition.
Height and weight at screening.
Abbreviations: SD = Standard deviation; ADHD = attention-deficit/hyperactivity disorder; ADHD-RS-IV = Attention-Deficit/Hyperactivity Disorder Rating Scale Version IV.
Drug exposure
The present study allowed for LDX dose titration in 10-mg intervals until an optimal dose was achieved. Overall, the mean (SD) exposure to LDX was 6.69 (1.30) weeks. The mean (SD) duration of exposure for the six dose levels ranged from 1.60 (1.45) weeks for the 20-mg dose to 2.23 (1.72) for the 30-mg dose. At study exit, the most commonly prescribed doses were between 30 and 50 mg/day. The dose of LDX received by subjects in the ITT population at study exit was as follows: 20 mg/day, n = 34; 30 mg/day, n = 71; 40 mg/day, n = 61; 50 mg/day, n = 70; 60 mg/day, n = 47; 70 mg/day, n = 33 (Fig. 2). Subject compliance with treatment regimens as assessed for each subject by a percentage of the number of capsules taken divided by the days of dosing was high with overall mean (SD) compliance of 98% (3.9) and ranging from 76.9% to 108.7%.
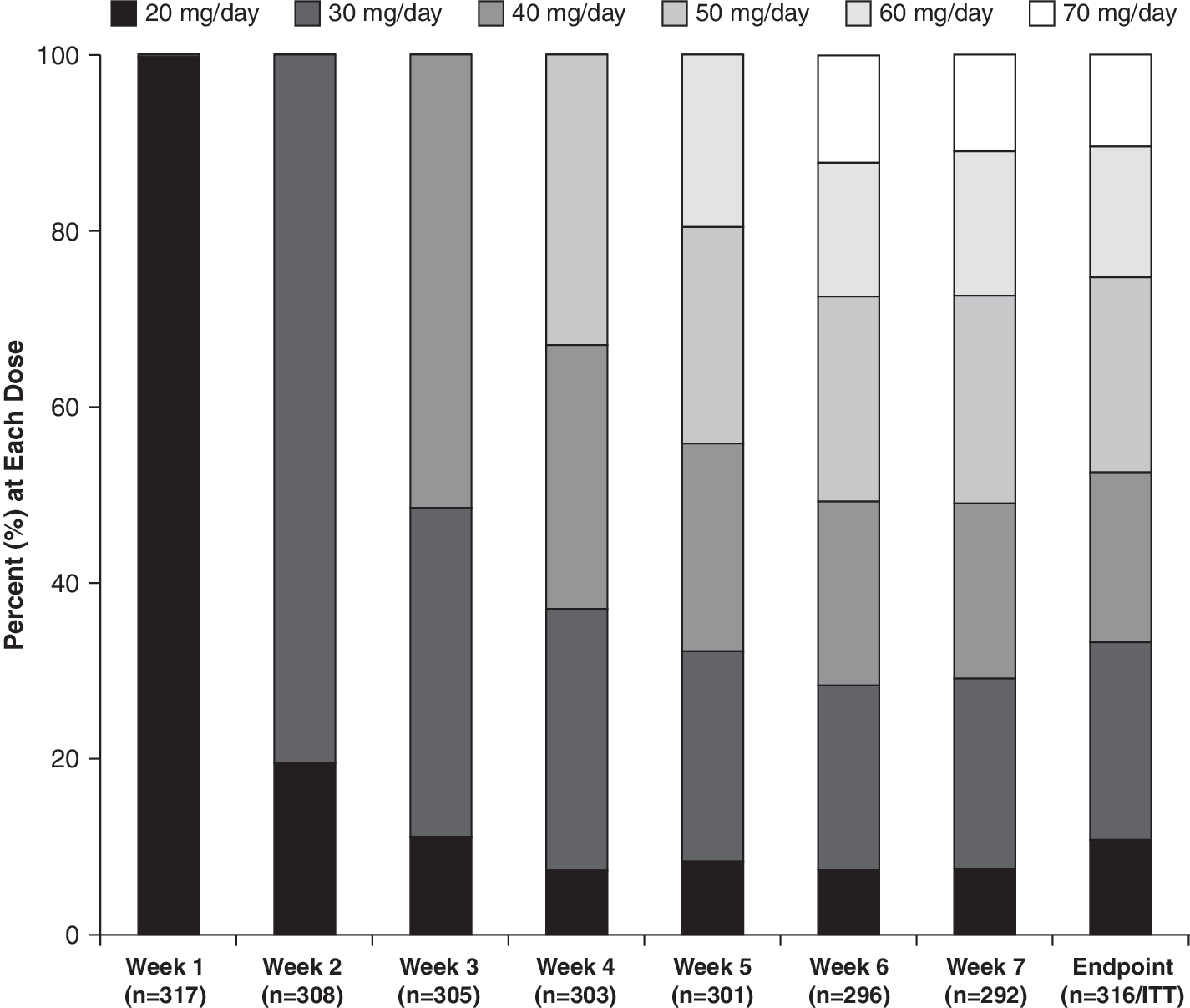
LDX dose distribution by week (safety population). Note that 1 subject started the study at 30 mg/day due to a dosing error. LDX = Lisdexamfetamine dimesylate; ITT = intention-to-treat.
Efficacy analyses
At baseline, the mean (SD) ADHD-RS-IV total score of the ITT population was 41.2 (7.0). At end point, defined as the last valid score after baseline, the mean (SD) ADHD-RS-IV total score was 12.6 (9.8). The mean (SD) change from baseline at end point was −28.6 (10.9) (p < 0.0001), a 69.3% average relative improvement. Moreover, the mean change from baseline in ADHD-RS-IV total score at each postbaseline visit was also significant (p < 0.0001) (Fig. 3). Improvements in ADHD-RS-IV total scores were similar between genders, with males demonstrating a mean (SD) decrease from baseline at end point of 28.3 (11.4) points, whereas females had a mean decrease of 29.2 (9.6) points. At end point, the mean (SD) change in ADHD-RS-IV total score for 20, 30, 40, 50, 60, and 70 mg/day LDX from baseline was −23.4 (13.5), −31.7 (9.8), −30.3 (8.7), −29.7 (9.1), −27.7 (11.4), and −23.4 (13.0), respectively (Fig. 4). Significant improvements in both the inattention (Fig. 5) and hyperactivity/impulsivity (Fig. 6) subscales of the ADHD-RS-IV from baseline were seen at each visit and at end point (p < 0.0001).
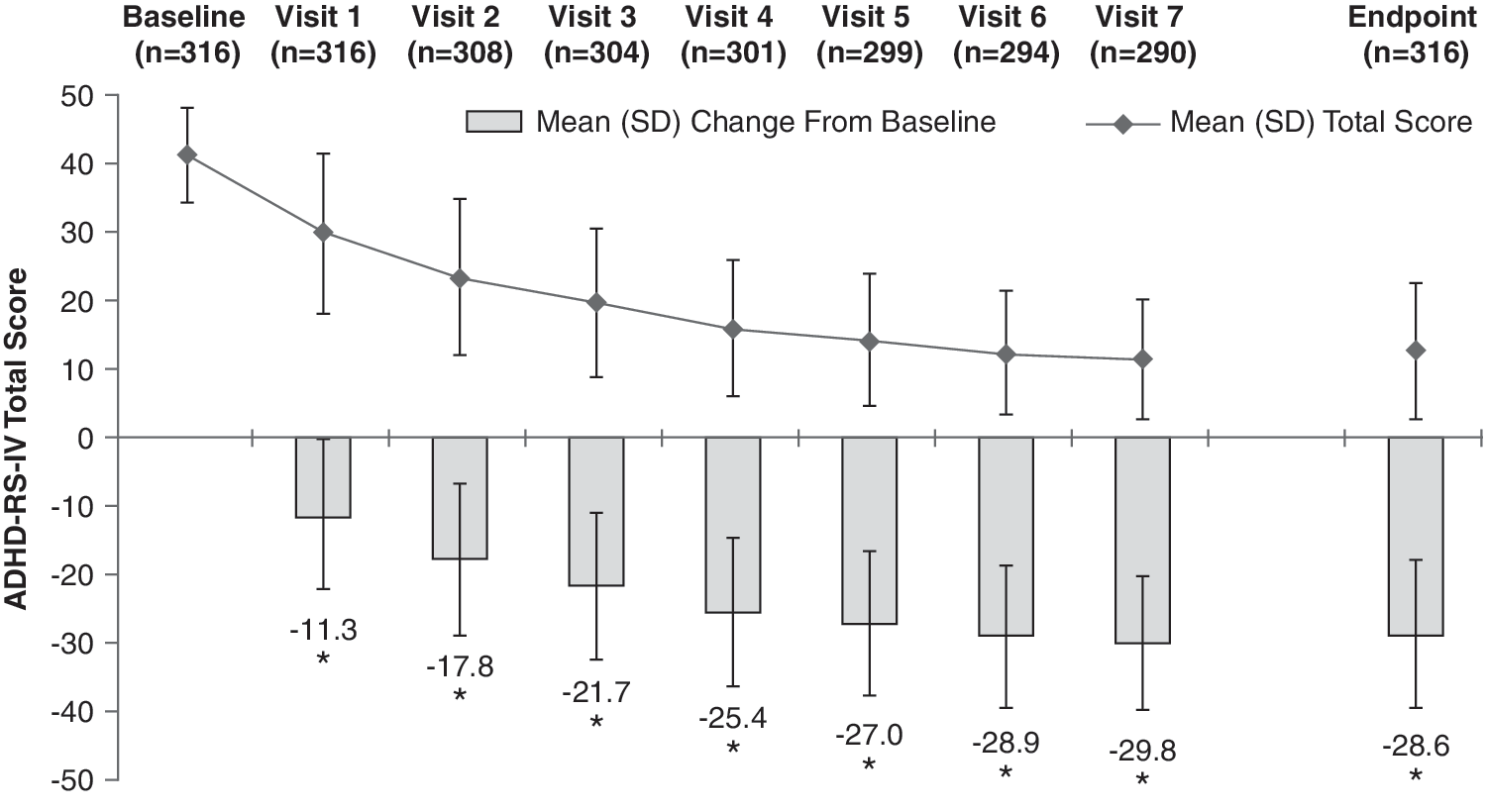
ADHD-RS-IV total score by visit and at end point (ITT population). End point defined as the last valid score obtained after baseline. (*) p < 0.0001 by one-sample t-test. ADHD-RS-IV = Attention-Deficit/Hyperactivity Disorder Rating Scale Version IV; ITT = intention-to-treat; LDX = lisdexamfetamine dimesylate; LOCF = last observation carried forward; SD = standard deviation.
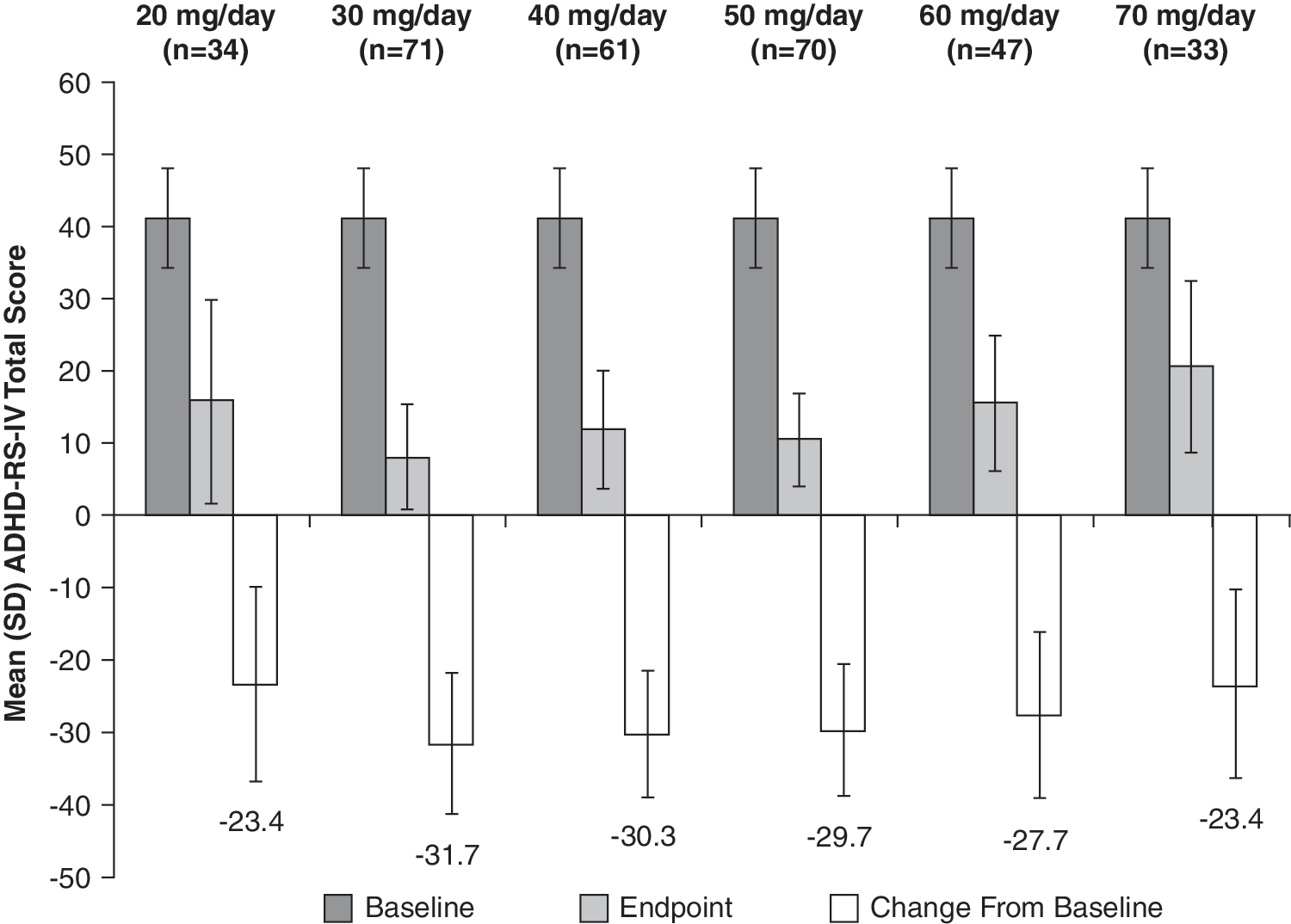
ADHD-RS-IV total score by LDX dose at end point (ITT population). End point was defined as the last valid score obtained after baseline. ADHD-RS-IV = Attention-Deficit/Hyperactivity Disorder Rating Scale Version IV; ITT = intention-to-treat; LDX = lisdexamfetamine dimesylate; SD = standard deviation.
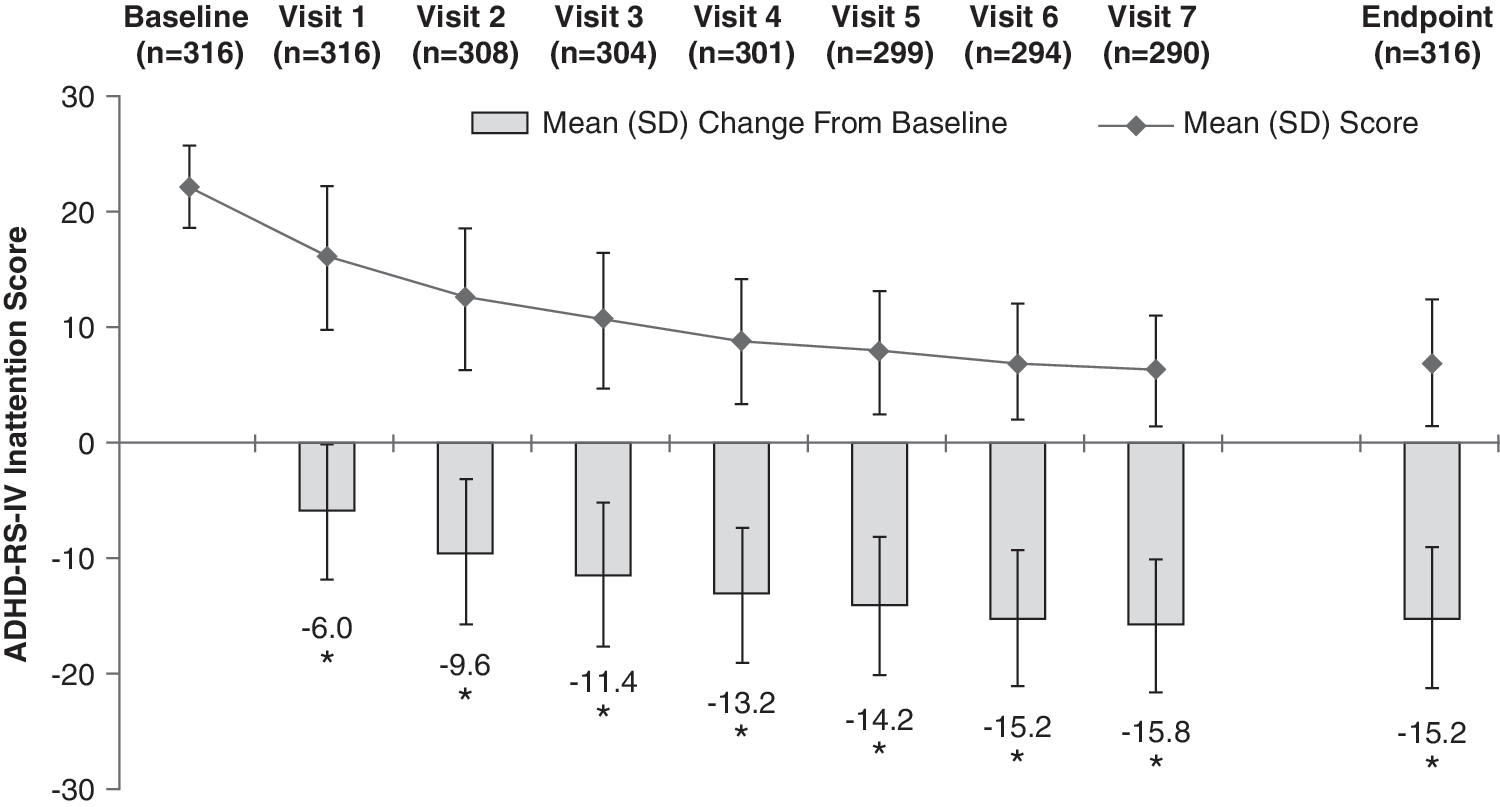
ADHD-RS-IV inattention score by visit and at end point (ITT population). End point was defined as the last valid score obtained after baseline. (*) p < 0.0001 by one-sample t-test. ADHD-RS-IV = Attention-Deficit/Hyperactivity Disorder Rating Scale Version IV; ITT = intention-to-treat; LDX = lisdexamfetamine dimesylate; SD = standard deviation.
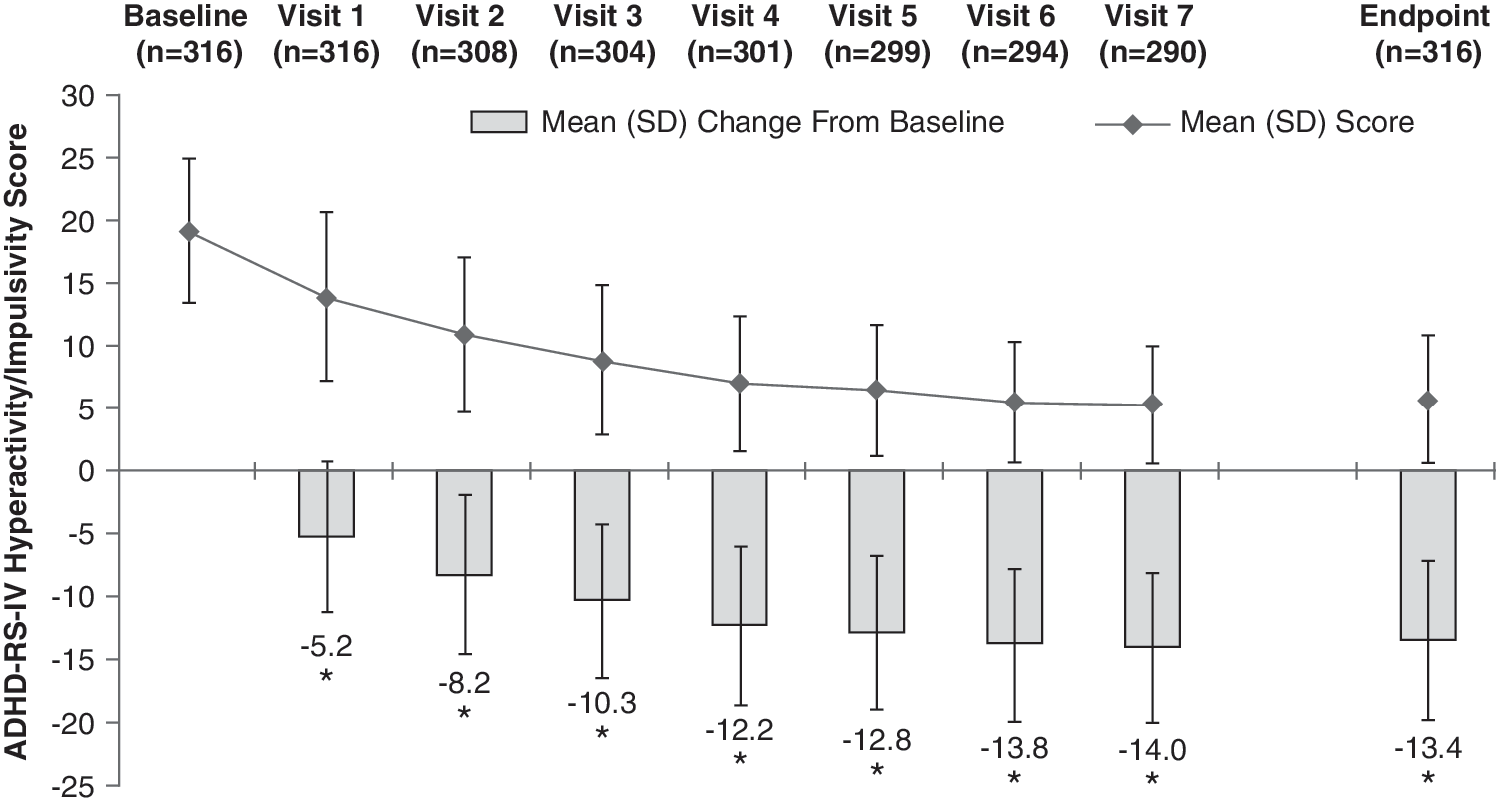
ADHD-RS-IV hyperactivity/impulsivity score by visit and at end point (ITT population). End point was defined as the last valid score obtained after baseline. (*) p < 0.0001 by one-sample t-test. ADHD-RS-IV = Attention-Deficit/Hyperactivity Disorder Rating Scale Version IV; ITT = intention-to-treat; LDX = lisdexamfetamine dimesylate; SD = standard deviation.
At baseline, the mean (SD) CGI-S score among patients in the ITT population was 4.7 (0.7). All patients were rated as having a severity of illness of 4 or greater with 41.8% (n = 132), 47.2% (n = 149), 9.8% (n = 31), and 1.3% (n = 4) of subjects assessed as moderately ill, markedly ill, severely ill, and among the most extremely ill subjects, respectively. At end point, the mean (SD) CGI-I score was 1.5 (0.8). Dichotomized CGI-I scores demonstrated that at end point, 89.9% of subjects were classified as improved (i.e., CGI-I of 1 or 2) (Table 2). Overall, the mean (SD) PGA score at end point was 1.8 (0.8). Using dichotomized scores, a substantial majority of patients (85.0%) were classified as being improved on LDX treatment as assessed by the PGA at end point (i.e., a score of 1 or 2) (Table 3).
End point is defined as the last valid assessment obtained after baseline.
Abbreviations: CGI-I = Clinical Global Impressions-Improvement; ITT = intention-to-treat.
End point is defined as the last valid assessment obtained after baseline.
Abbreviations: PGA = Parent Global Assessment; ITT = intention-to-treat.
Subjects in the ITT population had a mean (SD) baseline EESC total score of 63.4 (18.0). At the end of the study, the mean (SD) EESC total score for all subjects was 55.9 (17.7) and mean total scores were reduced (improved) at all dose levels (Fig. 7A). Overall, a statistically significant (p ≤ 0.0002) decrease (improvement) from baseline in EESC total and subscale (i.e., PE, EF, and EL) scores was observed for subjects treated with LDX at their last study visit (Fig. 7B), suggesting there was no mean worsening of emotional expression scores.
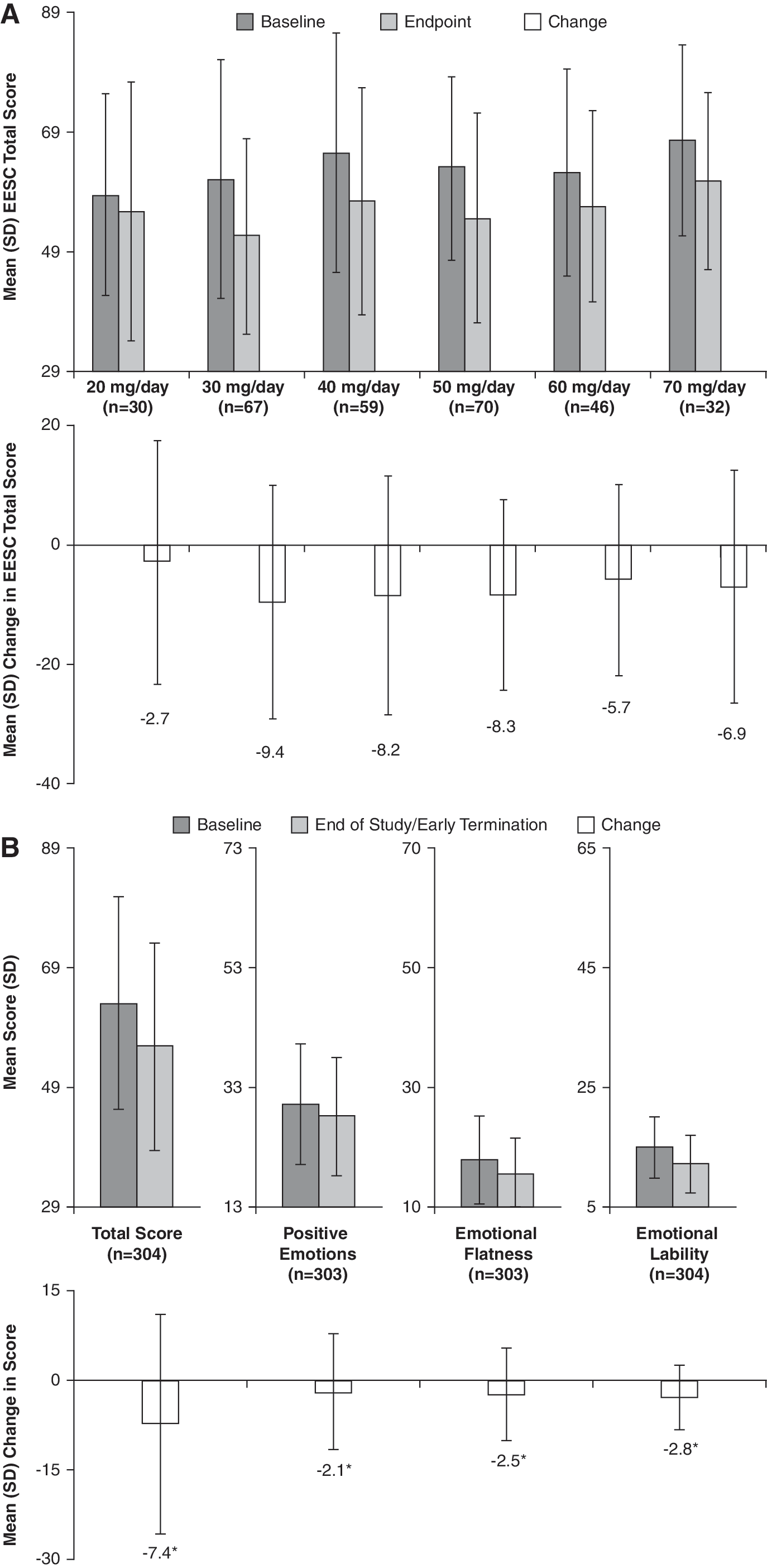
(
At baseline, the mean (SD) standardized T-scores for the BRIEF GEC among subjects in the ITT population was 74.0 (8.9). At the end of study, the mean (SD) GEC score for the ITT population was 56.1 (12.0). Subjects at all six dose levels demonstrated improvements in mean GEC score (Fig. 8A). Additionally, LDX treatment (all doses combined) was associated with significant improvement in scores of the BRIEF GEC as well as the two indices (i.e., MI and BRI) of the instrument (Fig. 8B) (p < 0.0001). At end point, the mean executive function T-scores were below a threshold (T-scores <65) that suggested the overall mean population did not have clinically significant impairment of executive function.
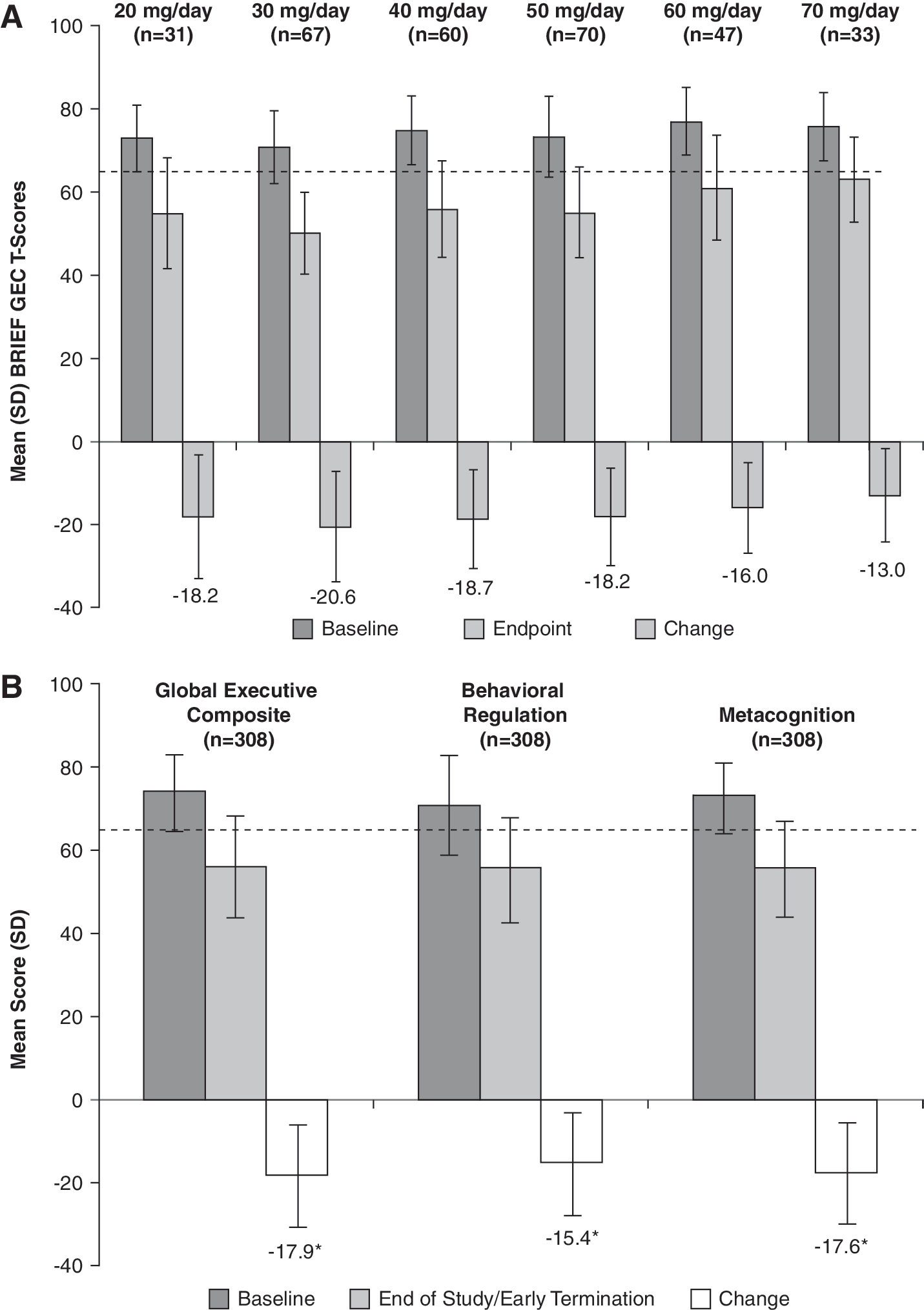
(
The MSQ asked parents to assess satisfaction with LDX treatment. When asked to rate their level of satisfaction with LDX, 76.0% of parents reported they were “very satisfied” with LDX (Table 4). Overall, as compared with their child's previous ADHD treatment, 31.2% of parents responded “not applicable” (e.g., child was not on medication previously), whereas 38.3% of parents rated LDX as “much better” and 19.5% rated LDX “better.” Only 2.3% of parents reported that LDX was “worse” than their child's previous treatment (Table 4). Finally, when queried about their desire to continue to use LDX, overall most parents (87.3%) said they would “absolutely” or “probably” continue to treat their child with LDX.
Abbreviations: MSQ = Medication Satisfaction Questionnaire; ADHD = attention-deficit/hyperactivity disorder; LDX = lisdexamfetamine dimesylate.
Safety analyses
Overall, 269 (84.9%) patients experienced one or more TEAEs. TEAEs reported by ≥10% of subjects were decreased appetite (43.2%), decreased weight (17.0%), irritability (16.1%), insomnia (16.1%), headache (13.9%), upper abdominal pain (13.2%), and initial insomnia (11.4%). The incidence of new TEAEs was highest at the 20-mg dose level (172 of 316 subjects, 54.4%) and lowest at the 70-mg dose level (9 of 37 subjects, 24.3%). Overall, 53.4% of subjects had TEAEs occurring during the first week of treatment, when each subject except one (secondary to a dosing error) was on the 20-mg dose.
Most TEAEs were mild or moderate in severity. Throughout the study, 7 (2.2%) patients reported a total of 9 severe TEAEs: Insomnia (n = 3), irritability (n = 2), decreased weight (n = 2), affect lability (n = 1), and initial insomnia (n = 1). All of these events were assessed to be related to treatment with LDX. Two serious TEAEs (STEAE) occurred during the present study. One event, syncope, occurred in a 12-year-old female with a prior history of intermittent syncope while receiving 60 mg/day LDX and was judged to be related to treatment. The patient completed the study and over the course of the study did not have any clinically significant ECG or vital sign abnormalities. The second STEAE was an episode of sinus arrest that occurred in a 7-year-old male while receiving 30 mg/day LDX. The patient was enrolled with an implanted loop recorder secondary to a prior episode of “near drowning,” but neither extensive prior cardiac evaluations, nor screening ECGs for the present study, revealed any abnormality. When the subject visited his cardiologist, a sinus pause lasting 6 seconds was noted on the recorder and the cardiologist recommended he undergo placement of an internal defibrillator. The subject was subsequently discontinued from the study. The investigator considered this event unrelated to the study drug.
Thirteen (4.1%) patients experiencing 16 events discontinued from the study secondary to TEAEs. All but one event (first-degree atrioventricular block) occurred after the first dose of LDX was administered and all events were considered treatment-emergent. The TEAEs leading to discontinuation included 3 cases of irritability, 2 cases each of aggression and affect lability, and 1 case each of initial insomnia, chest discomfort, ECG abnormality-QTc prolongation, tic, stomach discomfort, vomiting, blood pressure increase, and rash.
The mean (SD) baseline weight of subjects was 78.6 (22.5) pounds. At visit 7, subjects had a mean (SD) weight of 74.8 (21.6) pounds. Study participants exhibited a mean (SD) decrease in weight of 3.4 (4.0) pounds from baseline. Small increases in mean SBP, DBP, and pulse were seen throughout the study. At visit 7, the mean (SD) changes from baseline in vital signs were 0.9 (10.1) mmHg for SBP, 1.8 (9.3) mmHg for DBP, and 3.5 (12.7) beats per minute (bpm) for pulse. No patients met the outlier criteria for pulse (i.e., pulse ≤45 bpm or ≥130 bpm) during the present study. Sixteen subjects (5.0%) had SBP readings >120 mmHg and 8 subjects (2.5%) had DBP readings >80 mmHg at the screening or baseline visits. The frequency at which BP outlier criteria were met during postadministration of LDX is shown in Table 5. The range and distribution of change for weight and vital signs are illustrated in Fig. 9. The degree of dispersion for changes in weight and vital signs appears unremarkable.
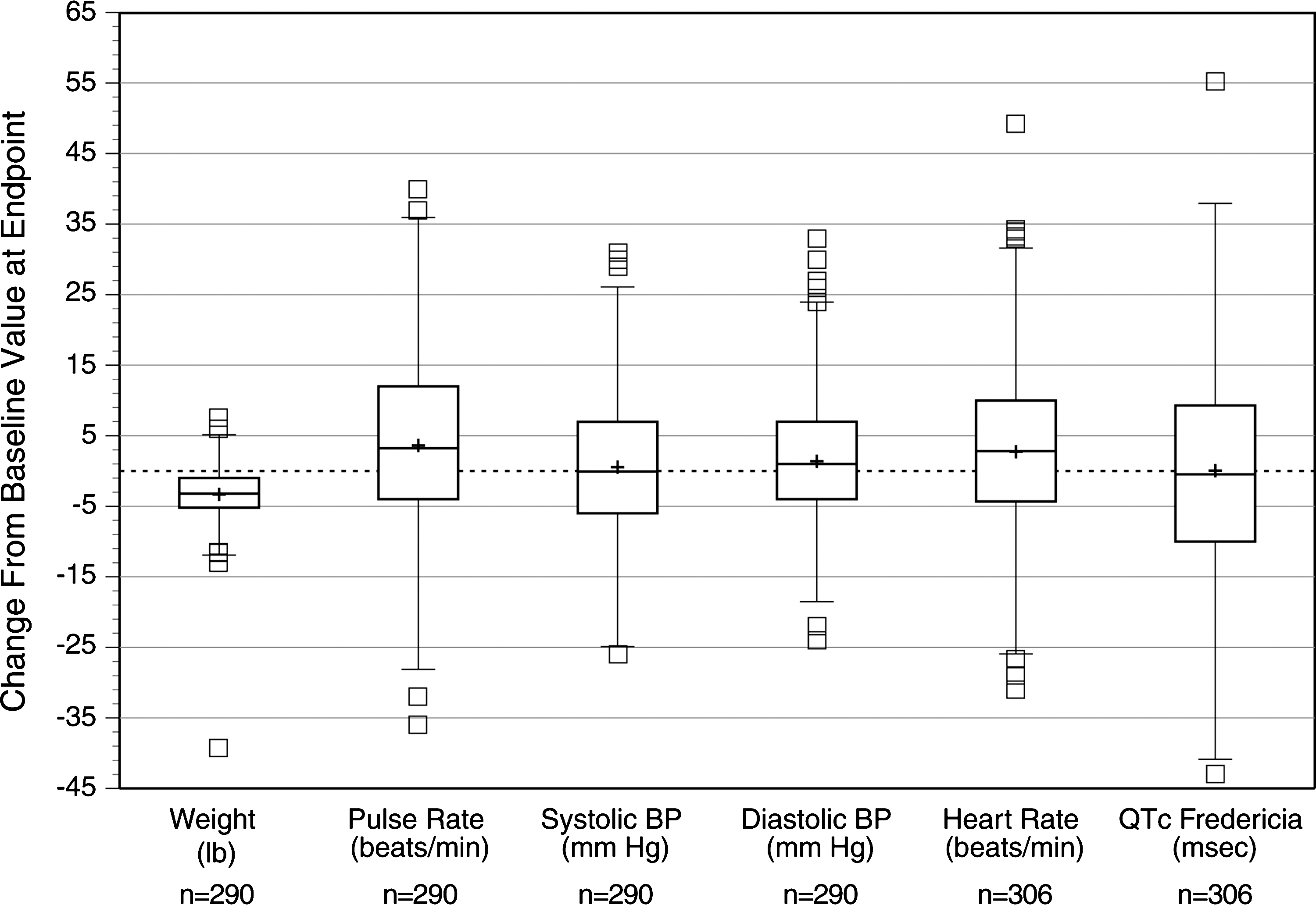
Descriptive statistics and distribution of body weight, vital signs, and QTcF interval. Note: Box and whisker plots illustrate mean (+sign within box), median (line through the center of box), 75th percentile (top line of box), 25th percentile (bottom line of box), 1.5 times the interquartile range (bars projecting up and down from the box), and outlier values falling outside 1.5 times the interquartile range (small boxes above and below box and whisker).
At the end-of-study/early termination visit, ECG recordings demonstrated a mean (SD) increase in heart rate of 2.8 (11.3) bpm. At the same assessment, subjects demonstrated a mean (SD) change from baseline of −4.1 (21.1) msec, 2.2 (17.5) msec, −0.1 (14.4) msec in QT, QTcB, and QTcF intervals, respectively. A total of 25 subjects met predefined criteria for ECG parameters of clinical interest at the end of study visit. One subject (0.3%) had a heart rate ≥130 bpm, while outlier criteria for PR interval (i.e., ≥180 msec), QRS interval (i.e., ≥100 msec), and QT interval (i.e., ≥450 msec) were met by 2 (0.6%), 5 (1.6%), and 0 subjects, respectively. Additionally, 9 (2.9%) subjects had a QTcB interval ≥450 msec. No subject had QTcF, QTcB, or QT intervals ≥480 msec. QTcB change from baseline ≥30 msec was observed in 14 (4.5%) subjects; this change was ≥60 msec in one of these subjects. QTcF change from baseline ≥30 msec occurred in 4 (1.3%) subjects; none of these subjects had a QTcF change from baseline to end of study of ≥60 msec. The range and distribution of change in heart rate and QTcF are illustrated in Fig. 9. The degree of dispersion for changes in heart rate and QTcF appear unremarkable.
Discussion
In this study, treatment with LDX in doses of 20–70 mg/day resulted in significant reductions of ADHD symptoms as assessed by the ADHD-RS-IV. Global improvements were observed as assessed by both the clinician-rated CGI-I and the parent-rated PGA. Teacher-rated assessments were not included in this study. By MSQ ratings, parents were generally satisfied with LDX, with most reporting that they were “very satisfied.” The majority of parents reported that LDX was better than their child's previous ADHD medication(s) and most reported that they would continue using LDX.
LDX was generally well tolerated, with most TEAEs being mild or moderate in severity. The incidence of new TEAEs was highest at the beginning of treatment (i.e., at the first dose strength of 20 mg) and generally declined thereafter. Overall, the TEAE profile demonstrated in this study was similar to that of previous studies of LDX and other stimulants (Biederman et al. 2007a; Biederman et al. 2007b; Findling et al. 2008). Although 1 patient experienced an STEAE of sinus arrest (judged not related to treatment), no clinically meaningful trends were observed for the ECG interval data, and the small mean increases observed for pulse and BP were consistent with stimulant therapy. The decrease in weight associated with LDX is consistent with other stimulants as well as with data from previous clinical trials of LDX (Spencer et al. 2006; Biederman et al. 2007b; Adderall XR [package insert]; Vyvanse [package insert])
The recently described EESC has been developed to be a useful measure of emotional expression in children with ADHD (Kratochvil et al. 2007; Perwien et al. 2008). In this study, it was found that subjects treated with optimized dosing of LDX demonstrated small, yet statistically significant, improvements in EESC total scores. Additionally, statistically significant improvements were observed in the positive emotion, emotional flatness, and emotional lability subscales. Although interpretation of these findings is limited by the lack of both a placebo group for comparison and normative data for the EESC, they constitute encouraging results suggesting that the treatment of children with ADHD with LDX does not negatively affect emotional expression overall. Additional data regarding the effect of LDX on emotional expression and regulation were captured by TEAE data. Overall, 7.3% (n = 23) of subjects were reported to exhibit affect lability whereas 3 subjects (0.9%) demonstrated a flat affect and 1 subject (0.3%) each reported blunted affect and apathy. Two patients (0.6%) reported mood swings. Tearfulness was reported by 4 patients (1.3%), whereas dysphoria and depressed mood were each reported by 1 subject (0.3%). Whether these TEAEs are attributable to treatment with LDX or represent symptomatology of underlying ADHD is unknown.
Deficits in executive functioning, as measured by neuropsychological tests, are more common in patients with ADHD compared with controls (Biederman et al. 2006). A broader definition of executive functioning, based on performance in everyday activities, has been used to develop the hypothesis that impairment of executive function is central to ADHD (Barkley 2003; Brown 2006). Deficits in executive functions as assessed by “real-world” activities have been demonstrated in children with ADHD (Lawrence et al. 2002; Lawrence et al. 2004). The BRIEF was designed to assess executive functioning using a behavior rating scale rather than neuropsychological testing, and several recent studies have provided evidence supporting the validity and utility of the BRIEF as a tool for evaluating executive function in subjects with ADHD (Gioia et al. 2002; Mares et al. 2007; McCandless et al. 2007). In the present study, LDX treatment was associated with statistically significant improvements in the overall GEC score as well as the BRI and MI scores of the BRIEF. During treatment with LDX, the mean BRIEF GEC, BRI, and MI T-scores at all dose levels were less than 65, a cutoff that is associated with potential clinical significance (Gioia et al. 2000; Mahone et al. 2007; McCandless et al. 2007). The finding that treatment with the long-acting prodrug LDX resulted in improvements in BRIEF scores provides support for the view that effective treatment of ADHD in children may be associated with improved executive function as perceived by their parents in the real world.
This is the first clinical trial to demonstrate effectiveness for LDX at doses ranging from 20 to 70 mg. Although not designed to provide dose–response information, the present study demonstrated that across a wide range of doses, subjects achieved reduction in ADHD symptomatology. Less than 15% of subjects required escalation up to 70 mg/day LDX during the trial. Furthermore, at week 1, when all but 1 patient was receiving 20 mg/day LDX, significant reductions in mean ADHD-RS-IV total score and the inattention and hyperactivity/impulsivity subscales were observed, suggesting that 20 mg may be an effective dose for some patients. Compared to forced-dose titration protocols, the dose-optimization design of this large clinical trial may more closely reflect the common prescribing practices of clinicians treating children with ADHD. The recently expanded range of doses of LDX will likely provide clinicians greater flexibility in dosing so as to achieve optimal efficacy and tolerability for a broad range of patients.
The present study has several limitations. Children with co-morbid psychiatric disease with significant symptoms and substantive medical illnesses were excluded from the study, which may limit the ability to extrapolate these results to the segment of the ADHD patient population with significant co-morbid disorders. Subjects with prior intolerable response to amphetamines were excluded from the study, which may affect interpretation of incidence rates of some TEAEs. The open-label nature of this study does not allow for a reference arm to which the results of patients on LDX can be compared. Additionally, the design of the present study allowed physicians to make treatment choices on the basis of response and tolerability judgments during the dose-optimization phase; however, doses could not be increased during the dose-maintenance phase, potentially leading to an underestimation of effectiveness. While with such an open-label, dose-optimization design it seems reasonable to assume that isolated potential instances of positive bias (e.g., desire to achieve maximal benefit) or negative bias (e.g., especially vigilant concern to avoid intolerable TEAEs) may have occurred, the extent to which each form of bias occurred cannot be ascertained with certainty. Open-label design, of course, limits interpretation of TEAE incidence rates because no untreated control is available for comparison. As a short-term study, conclusions regarding the long-term effectiveness and safety of LDX cannot be drawn from these results; however, long-term results with dosing up to 70 mg (the highest dose used in the current study) have demonstrated that LDX is generally safe, effective, and tolerable for up to 1 year of treatment (Findling et al. 2008).
Conclusions
The present study suggests that the newly approved dosages of LDX will offer clinicians greater flexibility in titrating LDX doses so as to maximize symptom improvement while minimizing TEAEs. LDX was generally well tolerated with a safety profile consistent with long-acting stimulant use in this population of children 6 to 12 years of age with ADHD. LDX was effective at reducing symptoms of hyperactivity/impulsivity and inattention as measured by the ADHD-RS-IV that, at end point, the mean score was below 18, suggesting symptomatic remission of the mean population (Steele et al. 2006). Most subjects were also assessed to be “much” or “very much” improved by CGI-I scores. These results are consistent with the large effect sizes noted in a previous trial (Biederman et al. 2007b). Effectiveness was consistently demonstrated across parent- and clinician-rated scales. Additionally, treatment resulted in improvement of parent-rated behaviors that may be related to executive function, as measured by BRIEF scores, without overall worsening of measures of emotional expression as measured by EESC mean score at end point. These findings support the effectiveness and safety of LDX treatment in children with ADHD.
Footnotes
Acknowledgments
Writing and editorial assistance was provided by Health Learning Systems, part of CommonHealth®, and funded by Shire Development Inc. In particular, we acknowledge the contribution of Michael Pucci, Ph.D., medical writer for Health Learning Systems, who, along with the expert contribution and approval of Robert L. Findling, M.D., Lawrence D. Ginsberg, M.D., Rakesh Jain, M.D., and Joseph Gao, Ph.D., developed the first draft of this manuscript.
Author Disclosures
Dr. Findling receives or has received research support, acted as a consultant and/or served on a speaker's bureau for Abbott, Addrenex, AstraZeneca, Biovail, Bristol-Myers Squibb, Forest, GlaxoSmithKline, Johnson & Johnson, KemPharm, Lilly, Neuropharm, Novartis, Organon, Otsuka, Pfizer, Sanofi-Aventis, Sepracore, Shire, Solvay, Supernus Pharmaceuticals, Validus, and Wyeth. Dr. Ginsberg receives or has received research support, acted as a consultant and/or served on a speaker's bureau for Abbott, Alkermes, AstraZeneca, Bristol-Myers Squibb, Cephalon, Cyberonics, Forest, GlaxoSmithKline, Janssen, Jazz, JDS Pharmaceuticals, Eli Lilly, McNeil, Neurocrine Biosciences, Neuropharm, New River Pharmaceuticals, Novartis, Organon, Ortho-McNeil, Otsuka, Pam Labs, Pfizer, Sanofi-Aventis, Schwarz, Sepracor, Shire, Takeda, UCB Pharma, Validus, and Wyeth. Dr. Jain has acted as a consultant and has received honoraria and/or travel reimbursement from Addrenex, Impax, Eli Lilly, Pfizer, Shire, and Takeda. Dr. Gao is a full-time employee and stock holder of Shire Development, Inc., Wayne, PA.
This study was supported by Shire Development Inc., Wayne, Pennsylvania.