
Abstract
Objective:
This 5-week, multicenter, double-blind, placebo-controlled, parallel-group investigation is the first fixed-dose study to evaluate efficacy and tolerability of three doses of (10, 20, or 30 mg, once daily [o.d.]) dexmethylphenidate hydrochloride (HCl) extended-release (d-MPH XR; Focalin® XR) across multiple settings to treat pediatric attention-deficit/hyperactivity disorder (ADHD).
Results:
ADHD pediatric outpatients (n = 253) diagnosed according to Diagnostic and Statistical Manual of Mental Disorders, 4th edition, criteria were randomized (1:1:1:1) to receive d-MPH XR (10, 20, or 30 mg o.d.) or placebo. Treatment with d-MPH XR significantly (p < 0.001) reduced the mean score (change from baseline) on Conners'-ADHD/DSM-IV Scales (CADS) as assessed by the teacher CADS-T (dose [mean];10 mg [18], 20 mg [16.9], 30 mg [20.7]) and parents, CADS-P (dose [mean];10 mg [15.8]; 20 mg [17.8]; 30 mg [20.5]) compared to placebo (mean CADS-T [5.7]; CADS-P [4.6]). A significant (p < 0.001) proportion of patients in the three d-MPH XR treatment groups showed improvement on the clinician-rated, Clinical Global Impressions–Improvement (CGI-I) scales (10 mg [73.8%]; 20 mg [71.2%]; 30 mg [77.2 %]) and severity ratings (CGI-S) compared to the placebo group (CGI-I, 22.2%). Adverse events were mild to moderate in severity and similar to previous observations for this class of neurostimulants.
Conclusion:
All three doses of d-MPH XR (10, 20, or 30 mg o.d), were significantly more effective than placebo in improving ADHD symptoms as confirmed by the teacher, parent and clinician. Additionally, d-MPH XR was well tolerated and demonstrated a consistent safety profile.
Introduction
The racemic mixture of d-threo- and l-threo-enantiomers of methylphenidate hydrochloride (d,l-MPH) has been the mainstay treatment for ADHD (Greenhill et al. 1999; Faraone et al. 2006). MPH acts by increasing the synaptic concentration of the neurotransmitters, norepinephrine and dopamine, whose neuromodulatory influence over behavior and cognition via the fronto-striato-cerebellar circuitry enables an amelioration of problems related to impulsivity and inattention (Berridge et al. 2006; Wilens 2006; Chamberlain et al. 2007).
Dexmethylphenidate hydrochloride Extended Release (d-MPH XR) is an orally administered, bimodal release, capsule formulation of the d-threo-enantiomer of racemic MPH. The maximum recommended dose of d-MPH XR for current users is 20 mg once daily (o.d.) (Focalin Prescribing Information; Novartis Pharma), and previous studies mainly investigating doses of ≥20 mg daily have indicated the drug to be significantly more effective than placebo in reducing symptoms of ADHD in children with a tolerable safety profile (Greenhill et al. 2006; Silva et al. 2006; Brams et al. 2008; Silva et al. 2008) and adults (Spencer et al. 2007). However, these studies recorded efficacy responses primarily in classroom settings and provided limited dose-specific efficacy and safety information.
This study is the first fixed-dose study in multiple settings, at home, school, and clinic, designed to investigate the efficacy and safety of three doses of d-MPH XR (10, 20, or 30 mg) administered o.d. when compared to placebo in treating pediatric patients meeting the Diagnostic and Statistical Manual of Mental Disorders, 4th edition (DSM-IV) (American Psychiatric Association 1994) criteria for ADHD. The assessments across multiple settings from this outcome report provide a unique perspective on the overall treatment effect of d-MPH XR in pediatric ADHD. Additionally, the study outcome would be useful in ascertaining the efficacy response of a lower dose (10 mg o.d.) and the tolerability profile of a higher dose (30 mg o.d.) relative to the standard dose of d-MPH XR (20 mg o.d.).
Methods
Patients
The study population consisted of children of either gender (aged 6–12 years) diagnosed with ADHD of any subtype as per Diagnostic and Statistical Manual of Mental Disorders, 4th edition, Text Revision (DSM-IV-TR) (American Psychiatric Association 2000) criteria (predominantly inattentive, predominantly hyperactive/impulsive or combined). Diagnosis was verified by the investigator based on a psychiatric examination and a semistructured diagnostic interview (the ADHD module of the Kiddie Schedule for Affective Disorders and Schizophrenia–Present and Lifetime Version, [K-SADS-PL]). (Kaufman et al., 2007) It was essential that patients attending school had the same teacher (English or math) for the entire duration of the study, who was willing and able to spend sufficient time with the patient to make valid weekly assessments reflecting the child's symptoms over the past week. Patients were eligible for screening only if they were either drug naïve or not treated with any MPH-related medication in the month prior to the study. Patients receiving psychological or behavioral therapies before the screening visit were considered eligible to participate, provided that the therapy had been ongoing for at least 3 months with the same therapist. Patients had to have an academic competence appropriate to their age and the following subscale total scores on the Conners' ADHD/DSM-IV Scales for teacher (CADS-T): For boys, baseline scores on the CADS-T subscale total were required to be ≥27 for those 6–8 years old, ≥24 for those 9–11 years old, or ≥19 for those 12 years old. For girls, the respective baseline cutoff scores on the CADS-T for the same age groups were ≥16, ≥13, or ≥12 (Conners 1997).
The exclusion criteria included home-schooled children, any medical condition that interfered with study assessments or that was not stable for at least 3 months before screening, clinically significant abnormalities detected during screening, family history of long-QT syndrome, current diagnosis or a history of cardiac abnormalities, seizures, psychiatric disorders such as schizophrenia, schizoaffective disorder, severe obsessive-compulsive disorder, conduct disorder, autism, chronic tic disorder, Tourette disorder, and any mood or anxiety disorder. Antidepressants, antipsychotics, herbal preparations with psychotropic effects, amphetamine-based medications, benzodiazepines, barbiturates, sedatives or hypnotics, monoamine oxidase inhibitors, and atomoxetine had to be stopped 1–4 weeks prior to randomization according to their half-lives. All concomitant medications that could interfere with the absorption, metabolism, and distribution of the study drug were excluded from start of screening until the end of all evaluations. Over-the-counter analgesics, short-term antibiotic treatment for minor infections, and any medication needed to treat adverse events (AEs) were allowed. Additionally patients who were judged by the investigator as likely to be noncompliant with study procedures, including those with a suspected history of substance abuse, or those living with a person diagnosed with a substance abuse disorder or whose parent or guardian was unable or unwilling to complete the Conners' ADHD/DSM-IV Scales for parents (CADS-P) were also excluded from the investigation.
Study design
This was a 5-week, multicenter, double-blind, randomized, placebo-controlled, four-arm, parallel-group trial (Fig. 1) in 253 pediatric outpatients with ADHD conducted across 34 centers in the United States. A sample size of 252 patients (63 per treatment group) was considered to provide 90% power to detect a treatment effect in any of the three active-treatment groups at the two-sided alpha level of 0.05 (treatment difference, 9.0 points between CADS-T and placebo; common standard deviation [SD], 13.5 points). Calculation of sample size was conducted using Bonferroni adjustment (Hochberg 1988). The institutional review boards or ethical review committees of each center approved the study, and written informed consent for participation was obtained from the parent or legal guardian of each child. Assent was obtained from children aged 7 years or older. The study followed good clinical practice guidelines and was in compliance with the Declaration of Helsinki.
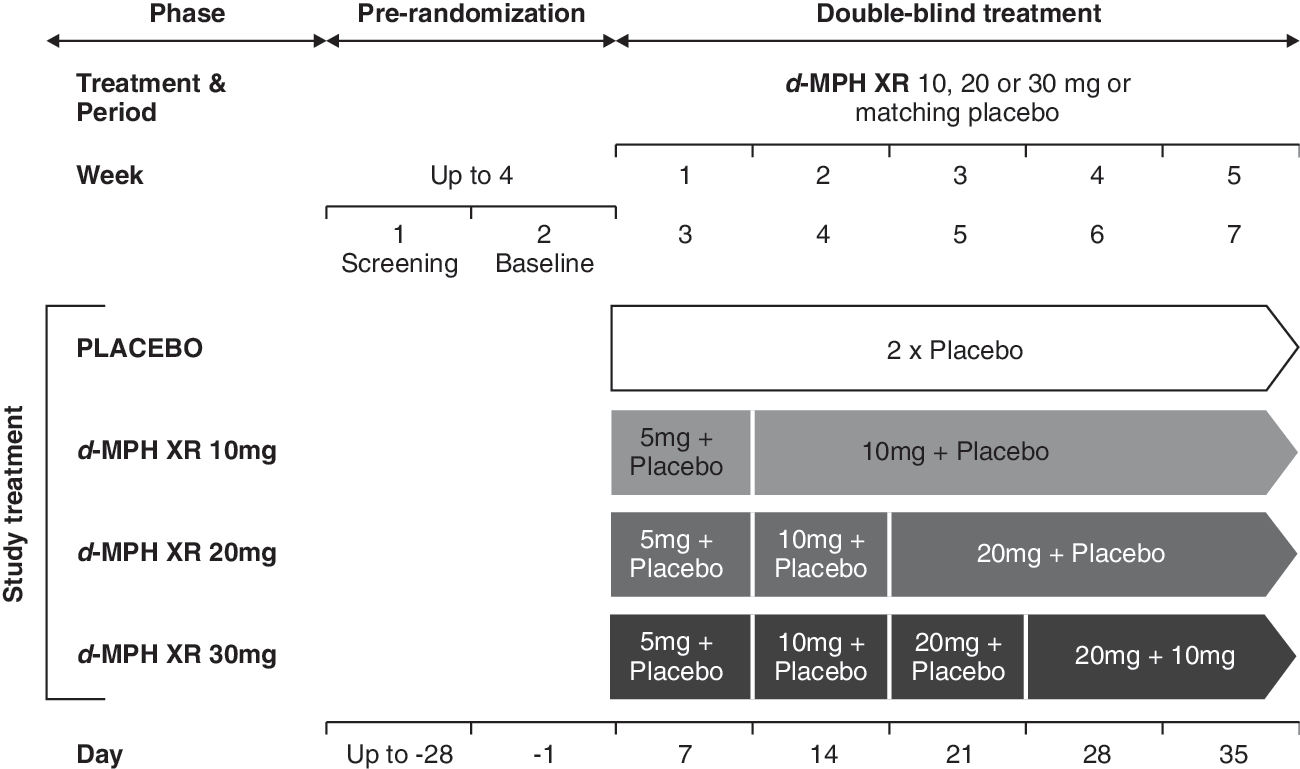
Study design. d-MPH XR, Dexmethylphenidate extended release.
The study was conducted in two phases: Prerandomization (screening) and double-blind treatment. During the screening phase (duration dependent on the half-life of any previous psychotropic medication), eligibility of patients was assessed by the application of inclusion/exclusion criteria and CADS-T score ratings.
At the end of screening, patients who met all inclusion/exclusion criteria were randomized (1:1:1:1) into one of the four treatment arms: d-MPH XR (10, 20, and 30 mg/day) or placebo. The drug was administered orally (two capsules) once daily in the morning by the parent/guardian.
The 5-week, double-blind treatment phase included a titration and a maintenance period. For the first week of treatment at baseline (visit 2), all patients randomized to active drug received d-MPH XR 5 mg daily. At visit 3, the dosage of active medication was increased to 10 mg/day. In the subsequent weeks, the dosage of 10 mg daily, was maintained in the patients randomized to the 10-mg dosage group, but increased in increments of 10 mg/week until those patients randomized to higher dosage groups reached their randomly assigned fixed dosage (20 or 30 mg/day). This meant that patients of the higher-dose groups (20 or 30 mg/day) received their assigned dose only in the third (20 mg/day) and fourth (30 mg/day) week, respectively. Randomization was performed using a validated system that automated the assignment of treatment arms to randomization numbers in the specified ratio.
Efficacy and safety assessments
Efficacy assessments were performed throughout the study by the parent, teacher, and investigator. The effectiveness of the drug was judged based on the alleviation of ADHD symptoms (inattention, impulsivity, or both) as rated by the teacher in school using the CADS-T, the parent or guardian at home using the CADS-P, and the clinician in the clinical setting by the Clinical Global Impressions–Improvement (CGI-I) and CGI–Severity (CGI-S) scales (Guy 1976).
The teacher's (math or English) assessment of hyperactivity and inattention symptoms on the CADS-T form was obtained before the baseline visit and once each week thereafter. The parent/guardian assessment was obtained weekly upon completion of the CADS-P. The CADS-P included the ADHD Index (12 items) and the DSM-IV subscale total (18 items). Both CADS-P and CADS-T had similar instructions for completion of the assessments.
The clinical investigator performed the CGI-S and CGI-I. The CGI-I assessed the overall change of illness relative to baseline (Guy 1976) and consisted of 7 ratings that ranged from 1 = very much improved to 7 = very much worse. The CGI-S rating indicated illness severity at each time point on a scale from “not at all ill” to “extremely ill.” CGI-I assessments were completed each week starting at visit 3 and compared current status to the study baseline (visit 2). CGI-S was performed at baseline and visit 7.
The primary efficacy variable for the present study was change from baseline to final visit in the DSM-IV subscale total score of the CADS-T after 5 weeks of double-blind treatment.
The secondary efficacy variables included change from baseline to final rating in CADS-P–DSM-IV subscale total score and improvement in rating at final visit on the clinician-rated CGI-S and CGI-I scales. Additional analyses included comparison of each d-MPH XR treatment to placebo over time for the primary efficacy variable and an estimation of treatment-by-center interaction using a mixed-effects analysis of covariance (ANCOVA) model.
The safety and tolerability of all three doses of d-MPH XR compared with placebo was also assessed. Safety assessments included regular monitoring and recording of AEs, serious adverse events (SAEs), vital signs, body weight, electrocardiogram (ECG), physical examination, hematology parameters, blood chemistry, and urinalysis. While vital signs were recorded in every visit, physical, laboratory and ECG evaluations were completed during the screening visit and final study visit. Weight was recorded at baseline and final visit. Notable value for weight loss was defined as having a decrease from baseline weight at study end of ≥7%.
Statistical analysis
The primary efficacy population was the intent-to-treat (ITT) population comprising all randomized patients who received at least one dose of study drug and had at least one postbaseline CADS-T assessment. Missing individual subscale scores and total subscale scores were imputed by using the last observation carried forward (LOCF) approach. The safety population consisted of all patients who had received at least one dose of study drug and had at least one postbaseline safety assessment. Patients were analyzed according to treatment received for all summaries based on the safety population.
Summary statistics (n, mean, SD, median, minimum, and maximum) were calculated for continuous variables and analysis of categorical variables was represented as frequencies and percentages. The primary statistical evaluation applied an ANCOVA on the change from baseline to final visit in the DSM-IV subscale total score of the CADS-T using the LOCF approach. Explanatory variables in the model included treatment group, pooled center, and the baseline DSM-IV subscale total score of the CADS-T.
The Hochberg procedure was applied to adjust for multiplicity because all three doses of d-MPH XR were simultaneously compared with placebo (Hochberg 1988). Tests of hypotheses were two-sided and were based on the three contrasts between each of the three d-MPH XR doses and placebo within the ANCOVA model, resulting in three p-values.
The CADS-P DSM-IV subscale total was analyzed using ANCOVA models similar to those applied in the primary efficacy analysis on the LOCF data set. The final CGI-I and CGI-S scale scores were analyzed using the Cochran–Mantel–Haenszel test with modified ridit scores and the pooled center as a stratification variable.
Evaluation of safety results was based on the frequency of AEs and on the number of laboratory values that fell outside of prespecified ranges.
Results
Patient disposition is presented in Fig. 2. Among 332 patients who were screened, 253 were randomized to the double-blind treatment phase to receive d-MPH XR 10 mg (n = 66), 20 mg (n = 62), 30 mg (n = 60), and placebo (n = 65). Failure to meet diagnostic/severity criteria (n = 28), withdrawal of consent (n = 16) for participation, and other reasons (n = 35) (such as unacceptable medical history or test result, etc.) led to the withdrawal of 79 patients during screening.
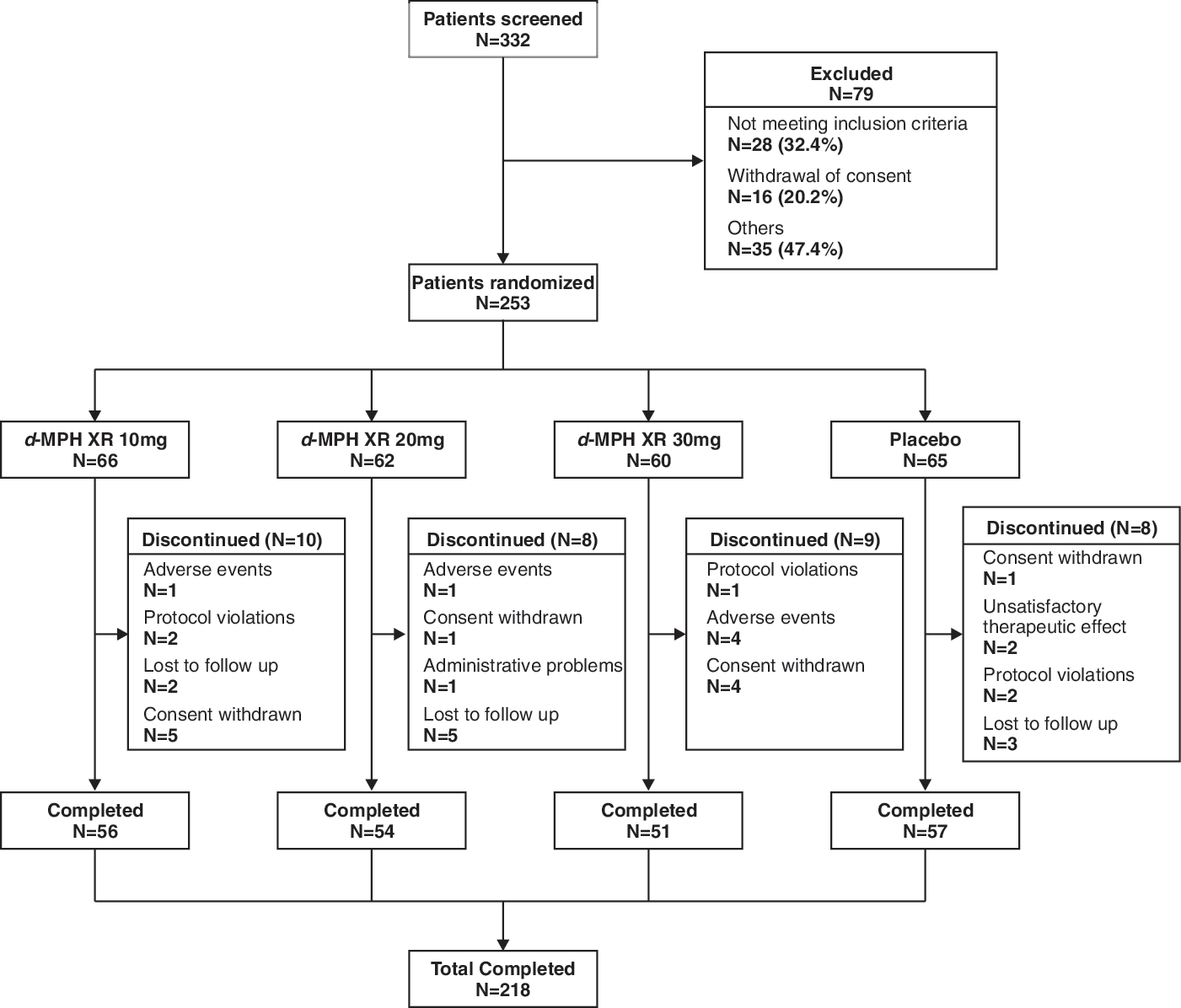
Patient disposition. d-MPH XR, Dexmethylphenidate extended release.
Among 253 randomized patients, the ITT population consisted of 240 patients (94.9%) and the safety population consisted of 245 patients (96.8%). Overall, 218 patients completed the study including 56 (84.8%), 54 (87.1%), 51 (85.0%), and 57 (87.7%) patients for d-MPH XR (10, 20, 30 mg) and placebo groups, respectively. There was no discontinuation due to unsatisfactory therapeutic effect of any d-MPH XR-treated patient from the double-blind treatment phase. The rates of discontinuation were similar in the d-MPH XR 10-mg (15.2%) and 30-mg (15.0%) groups, albeit slightly higher than the d-MPH XR 20-mg and placebo groups (12.9% and 12.3%, respectively). Across all treatment groups, the most common reasons for discontinuation were withdrawal of consent (d-MPH XR 10-mg group, 7.6%; d-MPH XR 30-mg group, 6.7%), loss of follow up (d-MPH XR 20-mg group, 8.1%; placebo group, 4.6%), and AEs (d-MPH XR 30-mg group, 6.7%).
Treatment groups were well balanced in relation to patient background characteristics and baseline demographics were comparable among treatment groups (Table 1) The mean age of the patient population was 8.7 years, and most patients were male (64.4%) and Caucasian (57.7%). The majority of patients (73.9%) were diagnosed with combined-type DSM-IV ADHD, the mean duration of ADHD symptoms was 3.9 years, and 69.2% of the patients were drug naïve.
Abbreviations: d-MPH XR = Dexmethylphenidate hydrochloride; AE = adverse events; SD = standard deviation; ADHD = attention-deficit/hyperactivity disorder.
The “All d-MPH XR” group had a higher proportion of patients with at least one protocol deviation (34.6%) compared to placebo (27.7%). The majority of protocol deviations were in the “study procedures category” (16.0% for “All d-MPH XR” and 13.8% for placebo). Deviations were mainly due to dose change or interruption during maintenance. Protocol violations due to use of barred psychotropic drugs during the study were more in the “All d-MPH XR” group (8.5%), in particular the 30-mg dose group (13.3%) than placebo (4.6%). The most frequently reported co-morbidities were under the following primary organ system classes: Respiratory, thoracic, and mediastinal disorders (26.9% “All d-MPH XR” vs. 38.1% placebo), immune system disorders (22.5% “All d-MPH XR” vs. 14.3% placebo), nervous system disorders (18.7% “All d-MPH XR” vs. 19.0% placebo), surgical and medical procedures (15.4% “All d-MPH XR” vs. 11.0% placebo), infections and infestations (14.3% in “All d-MPH XR” vs. 17.5 % in placebo), and skin and subcutaneous tissue disorders (11.0% in “All d-MPH XR” vs. 9.5% placebo).
The nervous system disorders (26.7%) and infections and infestations (18.3%) were most frequently reported in the d-MPH XR 20-mg group compared to all the other treatment groups. Surgical and medical procedures (20.7%) and skin and subcutaneous tissue disorders (15.5%) were predominantly reported in the d-MPH XR 30-mg group versus all other treatment groups.
The majority of patients in all treatment groups took at least one medication in the 30 days prior to the start of study (35.7% in “All d-MPH XR” group vs. 39.7% in placebo) and at least one concomitant medication or had a nondrug therapy after the start of the study (39.0% in “All d-MPH XR” group vs. 44.4% in placebo). The most common concomitant medications taken both prior to and after start of study were analgesics, antihistamines, or allergy medications, which are often seen in the age group (6–12 years old) recruited for the study.
Efficacy
The study compared the treatment effect of three different d-MPH XR doses to placebo but was not powered to show the difference in efficacy between these three active dose groups. Table 2 provides the mean scores (±SD) for the primary and secondary efficacy variables, CADS-T and CADS-P, respectively.
Abbreviations: d-MPH XR = Dexmethylphenidate hydrochloride; AE = adverse events; SD = standard deviation; CADS-T = Conners' ADHD/DSM-IV Scales for Teachers; CADS-P = Conners' ADHD/DSM-IV Scales for Parents.
As Fig. 3 indicates, patients in all three d-MPH XR-treated groups in the ITT population demonstrated a significant improvement as compared to placebo (all p < 0.001) with respect to the primary outcome measure, CADS-T, the teacher rating. The improvement in DSM-IV subscale total score (adjusted mean change from baseline) of the CADS-T was numerically greater in patients of the d-MPH XR 30-mg group (20.7) as compared with those in the 10-mg (18.0), 20-mg (16.9), and placebo groups (5.7).
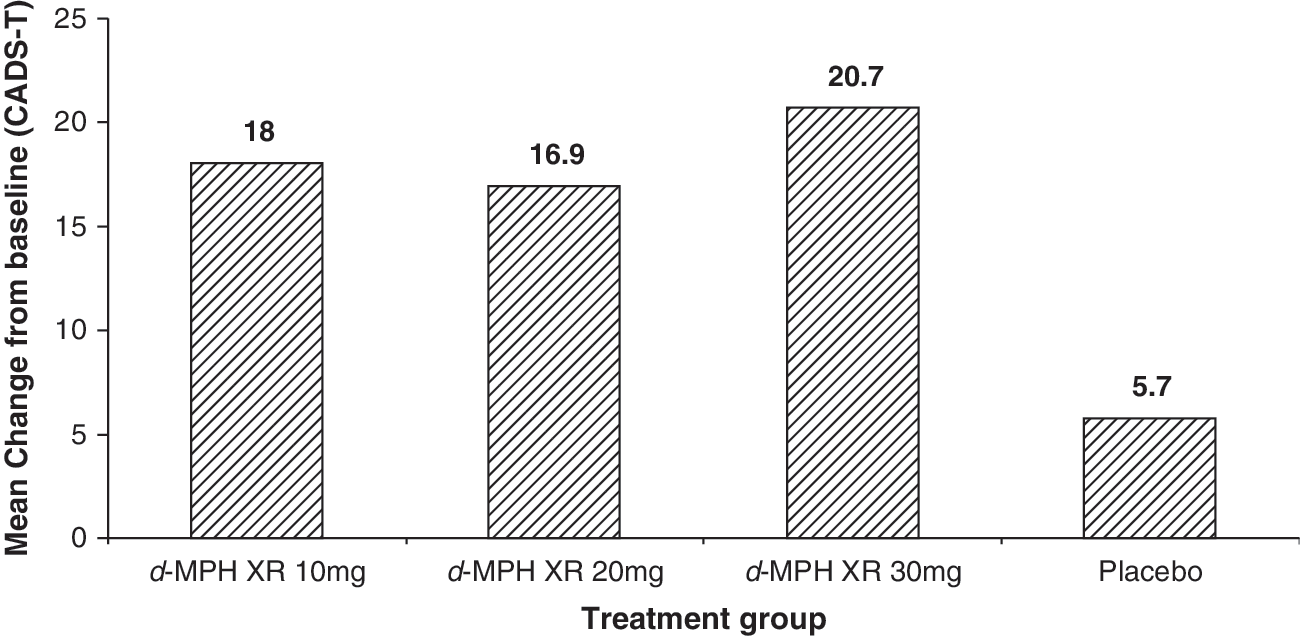
Change from baseline on the DSM-IV subscale total score of the CADS-T by treatment/last observation carried forward (intent-to-treat population). p < 0.001 based on the difference between each d-MPH XR group and placebo from the ANCOVA model. d-MPH XR, Dexmethylphenidate extended release; CADS-T, Conners' ADHD/DSM-IV Scale for Teacher; ADHD, attention-deficit/hyperactivity disorder; DSM-IV, Diagnostic and Statistical Manual of Mental Disorders, 4th edition; ANCOVA, analysis of covariance.
Additionally, a significant (all groups, p < 0.001) treatment effect over time was observed when each group was compared to placebo for the change from baseline on the DSM-IV total subscale score of the CADS-T per visit during the 5-week study: Overall treatment effect (95% confidence interval [CI]), 10 mg, 10.0 (6.6, 13.3); 20 mg, 8.2 (4.8, 11.6); and 30 mg, 11.5 (8.1, 14.9). No significant (p = 0.5723) indication for a treatment-by-center interaction was observed.
For the parent rating efficacy variable, CADS-P, the mean change in DSM-IV subscale total score (Fig. 4) showed significant improvement from baseline for all treatment groups at week 5/final visit compared to the placebo group (p < 0.001 for all three d-MPH XR groups) in the ITT population. The adjusted mean change from baseline was progressively higher for d-MPH XR 10-mg (15.8), d-MPH XR 20-mg (17.8), and d-MPH XR 30-mg (20.5) groups, and least in the placebo (4.6) group.

Change from baseline on the DSM-IV subscale total score of the CADS-P by treatment/last observation carried forward (intent-to-treat population). p < 0.001 based on the difference between each d-MPH XR group and placebo from the ANCOVA model. d-MPH XR, Dexmethylphenidate extended release; CADS-P, Conners' ADHD/DSM-IV Scale for Parent; ADHD, attention-deficit/hyperactivity disorder; DSM-IV, Diagnostic and Statistical Manual of Mental Disorders, 4th edition; ANCOVA, analysis of covariance.
The proportion of patients who were rated as “much improved” or “very much improved” on the CGI-I scales is presented in Fig. 5A. Patients in all the d-MPH XR treatment groups showed a statistically significant (p < 0.001) improvement on the CGI-I scale (73.8%, 45/61 for the 10-mg group; 71.2%, 42/59 for the 20-mg group; 77.2%, 44/57 for the 30-mg group) as compared with the placebo group (22.2%, 14/63). The assessment was missing for 1 patient in the d-MPH XR 20-mg group.

(
In addition to CGI-I, the efficacy measure, CGI-S was assessed at baseline and at the final visit (Fig. 5B). The distribution of the CGI-S ratings of each d-MPH XR treatment group was statistically significant better (p < 0.001) compared to the placebo group.
Safety
The overall incidence of AEs was generally higher in the d-MPH XR treatment groups than in the placebo group: 62.5% of patients in the d-MPH XR 10-mg group had at least one AE, 58.3% in the 20-mg group, and 70.7% in the 30-mg group versus 57.1% in the placebo group. The AEs that occurred at frequencies ≥2% in any treatment group are presented in Table 3. The most commonly reported individual AEs were upper abdominal pain (12.6% “All d-MPH XR” vs. 11.1% placebo), decreased appetite (12.1% “All d-MPH XR” vs. 4.8% placebo), and headache (12.1% “All d-MPH XR” vs. 12.7% placebo).The majority of AEs were reported in the d-MPH XR 30-mg treatment group, except for headache, which occurred mostly in the d-MPH XR 10-mg and 20-mg groups. Most of the AEs were mild and moderate in severity and categorized as psychiatric, metabolism and nutritional and gastrointestinal disorders. Among the psychiatric side effects, events such as insomnia and irritability occurred more often in patients assigned to the higher doses of d-MPH XR (20, 30 mg/day; Table 3).
Abbreviations: d-MPH XR = Dexmethylphenidate hydrochloride.
Five patients discontinued due to AEs (1 from d-MPH XR 10-mg group and 4 from d-MPH XR 30-mg group). Decreased appetite and insomnia were the most frequently reported AEs leading to discontinuation (1.1% for each of the two AEs in “all d-MPH XR” group).
There was one unexpected, drug-related, SAE reported in the d-MPH XR 30-mg group where the patient discontinued upon experiencing musculoskeletal stiffness and tactile hallucinations following drug administration. These symptoms resolved on discontinuation of the drug. No patient discontinued due to laboratory abnormalities; however, higher-than-normal values were observed for bilirubin (3.0%), potassium (5.1%), and serum alkaline phosphatase (3.2%) in the “all d-MPH XR” group, whereas none was reported in the placebo group. None of these abnormalities was reported as clinically significant by the investigators. No deaths were reported in this study.
Clinically notable changes in systolic blood pressure were observed in the d-MPH XR 30-mg group only (low value in 1 patient and high values in 2 patients). Three patients (2 high and one low) in the d-MPH XR 20-mg group and one patient in the placebo group (low level) exhibited clinically notable diastolic blood pressure readings.
Patients in the d-MPH XR treatment groups exhibited clinically notable changes in pulse rate. Low rates were seen in 2.4% of the patients in the “All d-MPH XR” group and in 5.3% of the patients in the placebo group, whereas high pulse rate was observed in 0.6% patients in the “All d-MPH XR” group only.
Only patients in the d-MPH XR treatment groups exhibited notable weight loss: 5.8%, 4.0%, and 7.7 % of patients in the d-MPH XR 10-mg, 20-mg, and 30-mg treatment groups, respectively. This weight loss is consistent with the adverse event profile known for this substance class.
Higher number of patients exhibited ECG abnormalities in the placebo group (5.3%) compared to the “d-MPH XR” group (3.8%). The most frequently reported ECG abnormality was intraventricular conduction defect (IVCD) in the “d-MPH XR” group (2.5%) compared to none in the placebo group. Occurrence of prolonged QTc was similar in both “d-MPH XR” group (1.3%) and the placebo group (1.8%). First-degree atrioventricular (AV) block was observed postbaseline in 1 patient each in the d-MPH XR 30-mg group and the placebo group. None of the abnormalities that were observed was considered clinically relevant by the investigator, and none were associated with SAEs.
Discussion
This was the first fixed-dose study to evaluate the benefits of three different doses of d-MPH XR in treating pediatric ADHD across multiple settings, as assessed by the teacher at school, parent at home, and the clinician. As noted by Collet et al. (2003), this method of multi-informant assessment is needed for the evaluation of ADHD medications because it enables improved clinical assessment, diagnostic determination, treatment monitoring, and, ultimately, accountability in practice.
The primary objective of this study was the evaluation by the teacher of the efficacy of once-daily administration of d-MPH XR (10 mg, 20 mg, and 30 mg) relative to placebo in improving control of pediatric ADHD symptoms on the CADS-T DSM-IV subscale total score. All three doses of d-MPH XR were statistically significantly better than placebo in improving the CADS-T DSM-IV subscale total score as assessed by the teachers from baseline to final visit, indicating that all doses of d-MPH XR studied exert a significant treatment effect in pediatric ADHD patients at school. Moreover, this significant treatment effect was observed over time for all three doses indicating a sustained efficacy. There was also no evidence of any influence of center over treatment effect.
The efficacy of 10, 20, and 30 mg of d-MPH XR relative to placebo in controlling ADHD symptoms was also rated by the parents (CADS-P). For the parent-rated CADS-P, the change from baseline to final visit between every d-MPH XR treatment group and placebo for the subscale total score was similarly statistically significant (p < 0.001). In addition, the clinician-rated CGI-I and CGI-S scores were also statistically significant for each d-MPH XR group (p < 0.001 for both ratings) as compared to placebo. These three different perspectives have confirmed the consistent performance and efficacy of d-MPH XR relative to placebo in alleviating the symptoms of pediatric ADHD.
Although the study was not powered to assess differences among active treatment groups, present findings indicate multiple doses (10 mg, 20 mg, or 30 mg) of d-MPH XR are more effective than placebo. A numerically greater improvement was observed for the patients in the d-MPH XR 30-mg group than for those in the 10- and 20-mg groups in relation to the CADS-T and CADS-P scores. Although optimal dosing cannot be determined based on the findings from this study, observations from previous studies have indicated that the treatment effect of MPH on the attention-based functions and behavior symptoms of pediatric ADHD is exerted in a dose-dependent manner and also depends on the subtype of ADHD (Sunohara et al. 1999; Markowitz and Patrick 2001; Stein et al. 2003; Konrad et al. 2004). Further studies are needed to investigate the dose-effect relationship in subgroups of patients, with different degrees of severity of the disease, and other factors such as age, gender, and weight.
In general, the efficacy of d-MPH XR as shown in this study is consistent with those observed in previous studies with either a flexible-dose design or assessed in a classroom setting. A 7-week, flexible-dose design study in 103 children and adolescents used the same outcome measures as the present study (CADS-T, CADS-P, and CGI-I) to judge the efficacy and tolerability of 5- to 30-mg/day doses of d-MPH XR in treating pediatric ADHD (Greenhill et al. 2006). Results showed that patients on d-MPH XR demonstrated an overall significant (p < 0.001) mean change from baseline for the CADS-T, CADS-P, and CGI-I scores as compared to placebo. Other studies in the classroom setting demonstrated the 20-mg/day dose of d-MPH-XR to offer significant, rapid (as early as 0.5 hours) and sustained efficacy for up to 12 hours postdose compared to placebo (Silva et al. 2006; Brams et al. 2008; Silva et al. 2008).
d-MPH XR belongs to a class of medications, the central nervous system (CNS) stimulants, which have, a well-established safety profile. Previous reports indicate that CNS stimulants cause AEs, including abdominal pain, loss of appetite, nausea, vomiting, somnolence, dry mouth, and emotional liability (Markowitz and Patrick 2001; Wigal et al. 2004). The AEs reported in this study were typical of those exhibited by drugs of this class. Psychiatric AEs occurred more often in patients treated with the higher doses of d-MPH XR. Because the effect of dose with regard to subtype and severity of ADHD was not investigated in this study, we cannot speculate regarding this finding.
One unexpected safety issue was reported in the d-MPH XR 30-mg treatment group, which led to the discontinuation of the patient from the study. The SAE included musculoskeletal stiffness and tactile hallucination. No deaths were reported in this study, and no patient discontinued treatment due to either lack of therapeutic efficacy or due to laboratory abnormalities, thereby indicating that all three doses of d-MPH XR were more effective than placebo in the treatment of pediatric ADHD and also well tolerated.
Limitations
Because this was a forced-dose titration study, some subjects may not have been treated with their optimal dose of d-MPH XR. Importantly, this study was not powered to assess the differences in treatment effects within the d-MPH XR groups; hence, an evaluation of the best dosage could not be made. Finally, although the study covers multiple settings, the short, 5-week, duration does not allow for extrapolation of these findings to long-term clinical ADHD outcomes.
Conclusion
This first fixed-dose study in multiple settings found once-daily administration of d-MPH XR (10, 20, and 30 mg) to be efficacious and safe in the treatment of pediatric ADHD symptoms, eliciting comparable positive therapeutic responses for all assessed doses when compared with placebo. The teacher, parent or guardian, and clinician consistently confirmed the efficacy of d-MPH XR and the suitability of a once-daily dosing regimen as indicated by the CADS-T and CADS-P subscale total scores and the CGI-I and CGI-S ratings. All ratings were statistically and clinically significant in favor of d-MPH XR versus placebo. Additionally, the safety profile of d-MPH XR was consistent with previously known profiles of neurostimulants.
Information from the present study is of relevance to clinicians and families of children afflicted with ADHD. Multiple perspectives—those of parent, teacher, and clinician—independently affirm the efficacy and safety of three different doses of once-daily d-MPH XR (10, 20, or 30 mg) in alleviating the impairing symptoms of ADHD. Additionally, the extended-release form of the drug could also improve patient compliance, because a once-daily administration can be done at home with no interruptions in school routine, making it convenient for both patients and caregivers.
Footnotes
Disclosures
Dr. Childress receives research support from, is a speaker for, or is a consultant of the following sources: Novartis Pharmaceuticals, Bristol-Myers Squibb Co., Abbott Laboratories, Shire, Astra-Zeneca, Lilly, Johnson & Johnson, Neuropharm, Somerset. Dr. Thomas Spencer receives research support from, is a speaker for, or is on the Advisory Board of the following sources: Shire Laboratories, Inc., Eli Lilly & Company, Glaxo-Smith Kline, Janssen Pharmaceutical, McNeil Pharmaceutical, Novartis Pharmaceuticals, Cephalon, Pfizer, and the National Institute of Mental Health. Dr. Lopez has been a consultant to, served on the speakers' bureaus of, and/or participated in research contracts with: Celltech-Medeva, Cephalon, Eli Lilly, New River Pharmaceutical, Noven, Novartis Pharmaceuticals, Shire Pharmaceutical, GlaxoSmithKline, and BMS. Ms. Gerstner, Ms. Thulasiraman, Dr. Muniz, and Dr. Post are employees of Novartis.
Acknowledgments
The authors would like to thank Dr. Anupama Shrinivasan (Novartis Pharma) for her help in the development of the manuscript.