
Abstract
Introduction:
Atomoxetine has been proposed to be effective for treating co-morbid attention-deficit/hyperactivity disorder (ADHD) in children with bipolar disorder (BPD) without destabilizing mood. We conducted an 8-week, open label study to study the efficacy and tolerability of adjunct atomoxetine in euthymic children and adolescents with BPD and ADHD.
Methods:
We evaluated 12 youth aged 6–17 years (mean = 11.3 years; 7 males) with a diagnosis of BPD I or II and ADHD. Subjects were euthymic at baseline and taking at least one mood stabilizer or antipsychotic. Primary outcome measure was the ADHD Rating Scale-IV (ADHD-RS-IV) (response = 25% decrease; remission = 40% decrease). Secondary outcome measures were change in Young Mania Rating Scale (YMRS) and Children's Depression Rating Scale (CDRS).
Results:
In primary outcome criteria, 8 (67%) were responders and 6 (50%) were remitters by ADHD-RS criteria. There was a significant decrease in ADHD-RS scores over the study (p < 0.0001; Cohen d = 2.18, effect size = 0.73). YMRS and CDRS scores did not change significantly from baseline to week 8. No subjects experienced a manic or mixed episode during the study, but 2 subjects were discontinued early due to worsening of mood symptoms.
Conclusions:
We found atomoxetine to be efficacious in treating symptoms of ADHD in children and adolescents with BPD taking mood stabilizers or antipsychotics. It is unclear whether symptomatic worsening of 2 subjects was due to atomoxetine or the natural course of illness. Placebo-controlled studies are needed to clarify the role of atomoxetine in this population.
Introduction
Atomoxetine is a Food and Drug Administration (FDA)-approved nonstimulant medication shown to be effective in the treatment of ADHD for children (Michelson et al. 2001; Michelson et al. 2002) and that may have promise for the treatment of ADHD symptoms in patients with BPD. Because it is not a stimulant, it would theoretically not have the same dangers as stimulants for inducing mania. Furthermore, clinicians may also be concerned regarding the possibility of inducing psychosis by stimulants and/or the potential for abuse of stimulants, and, as such, atomoxetine may not carry these same concerns, although empirical data in this area are lacking. Previously, we found adjunctive atomoxetine to be safe and efficacious in a retrospective case series of 7 children and adolescents with BPD and ADHD (Hah and Chang 2005). Therefore, we wished to study the effects of atomoxetine in this population prospectively in an open-label, 8-week trial.
Methods
This was an 8-week, open-label trial of atomoxetine added to mood stabilizers or antipsychotics in children and adolescents aged 6–17 years with BPD and ADHD. Informed written and verbal consent was obtained from the parent or legal guardian of the subjects, and written and verbal assent was obtained from the subjects. The protocol was approved by the Stanford University Panel on Human Subjects in Medical Research.
Inclusion criteria were: Males or females, 6–17 years old, with a diagnosis of BPD I or II by the Washington University in St. Louis Kiddie Schedule for Affective Disorders and Schizophrenia (WASH-U-KSADS) (Geller et al. 1996), and an ADHD-Rating Scale (ADHD-RS) score of at least national mean + 1.5 standard deviations (SD) for age and gender groups (DuPaul et al. 1998). Subjects were all outpatients. Co-morbid conditions of oppositional defiant disorder, separation anxiety, social phobia, and generalized anxiety disorder were allowed. Subjects were also required to be euthymic at baseline, as defined by at least 3 consecutive weeks of not meeting criteria for a hypomanic, manic, mixed, or depressive episode. During these three weeks, Young Mania Rating Scale (YMRS) scores needed to be <15, Children's Depression Rating Scale–Revised (CDRS) scores <35, and Clinical Global Impressions–Bipolar–Severity (CGI-BP-S) scores <3. Subjects had to be taking at least one mood stabilizer or antipsychotic, and no dose changes could be made within 3 weeks of baseline or during the study. Exclusion criteria included: Prior exposure to atomoxetine, substance dependence or use within 3 months prior to study, acute active suicidal ideation (i.e., plan or intent) or a recent (within 6 months) suicide attempt, a diagnosis of autism or Asperger's syndrome, mental retardation (suspected intelligence quotient [IQ] <80), a concomitant seizure disorder, or a current or history of hypertension.
We used Diagnostic and Statistical Manual of Mental Disorders, 4th edition (DSM-IV) (American Psychiatric Association 1994) definitions of BPD I and II. BPD I required a full manic episode (at least 7 days of elevated, irritable, or expansive mood, with three concurrent “B” symptoms of mania or four if the mood was irritable and not elevated). BPD II required a hypomanic episode (4–6 days of meeting symptom criteria for a manic episode), as well as a history of a major depressive episode.
Physical exams, including height and weight assessment, vitals, and an electrocardiogram (ECG) were performed at baseline and at week 8. Laboratories, including complete blood panel (CBC), renal panel, thyroid-stimulating hormone (TSH), β-human chorionic gonadotropin (β-hCG), liver function tests, and mood stabilizer serum levels, were also obtained at baseline and at week 8. At baseline, atomoxetine was begun at a dose closest to 0.5 mg/kg, increased at week 2 to 0.8 mg/kg, and increased thereafter to reach a target dose of 1.0 mg/kg, according to clinical response and adverse effects. Capsules were at fixed doses of 10 mg, 20 mg, or 40 mg, so a dose using a combination of these capsules closest to the calculated dose was used. The maximum dose allowed was 1.2 mg/kg.
Statistical analyses
The rating scales used weekly were the ADHD-RS, CDRS, YMRS, CGI-BP-S, and the Clinical Global Impressions–Bipolar–Improvement (CGI-BP-I). The primary outcome measure was response, as defined by a 25% decrease in ADHD-RS score at week 8. This level of response has previously been used for studies of atomoxetine in pediatric ADHD (Michelson, et al. 2001; Michelson, et al. 2002; Spencer et al. 2002). Remission was considered a 40% decrease in ADHD-RS score. Secondary outcome measures were changes in YMRS, CDRS, and ADHD-RS scores. We analyzed all data on our intent to treat (ITT) population, using SPSS 11.0 for the Macintosh. We used a last observation carried forward (LOCF) approach and analyses of variance (ANOVAs) for the analysis of outcome data (ADHD-RS, YMRS, CDRS). Paired t-tests were used to compare baseline and termination weight and vital sign changes.
Results
Twenty-seven subjects were screened by phone; 17 of those were enrolled, 5 were additional screen fails, and 12 received study drug (Fig. 1). We performed statistical evaluation on the ITT population (12 patients who took at least one dose of atomoxetine), whose age range was 6–14 years (mean age = 11.3 years; 7 males). The mean final dose of atomoxetine was 59.2 mg/day. Eight out of 12 (67%) of subjects were taking atypical antipsychotics, 9/12 (75%) anticonvulsants, and 2/12 (17%) were taking lithium. Eight out of 12 (67%) were responders and 6/12 (50%) were remitters by ADHD-RS criteria. There was a significant decrease in ADHD-RS scores over the study (t = 5.2, degrees of freedom [df] = 11, p < 0.0001; Cohen d = 2.18, effect size = 0.73) (Table 1). Eleven subjects (92%) experienced a decrease in their ADHD-RS score with a mean decrease of 16.9 ± 10.9), or a 43% decrease from the mean baseline ADHD-RS score of 39. YMRS and CDRS scores did not change significantly from baseline to termination (YMRS, −0.5 ± 5.5, p = 0.72; CDRS, −0.7 ± 6.7, p = 0.71) (Table 1). Weekly ADHD-RS, YMRS, and CDRS scores are presented in Figs. 2 and 3; week 8 scores differ from termination scores due to early termination of 2 subjects. One subject terminated due to hypomanic symptoms at week 4 and one to suicidal ideation at week 2. Two subjects (17%) were considered “much improved” in their overall bipolar symptomatology (CGI-BP-I = 2), 3 (25%) were “minimally improved” (CGI-BP-I = 3), 5 (42%) were “no change” (CGI-BP-I = 4), 1 (8%) was “minimally worse” (CGI-BP-I = 5), and 1 (8%) was “much worse” (CGI-BP-I = 6).
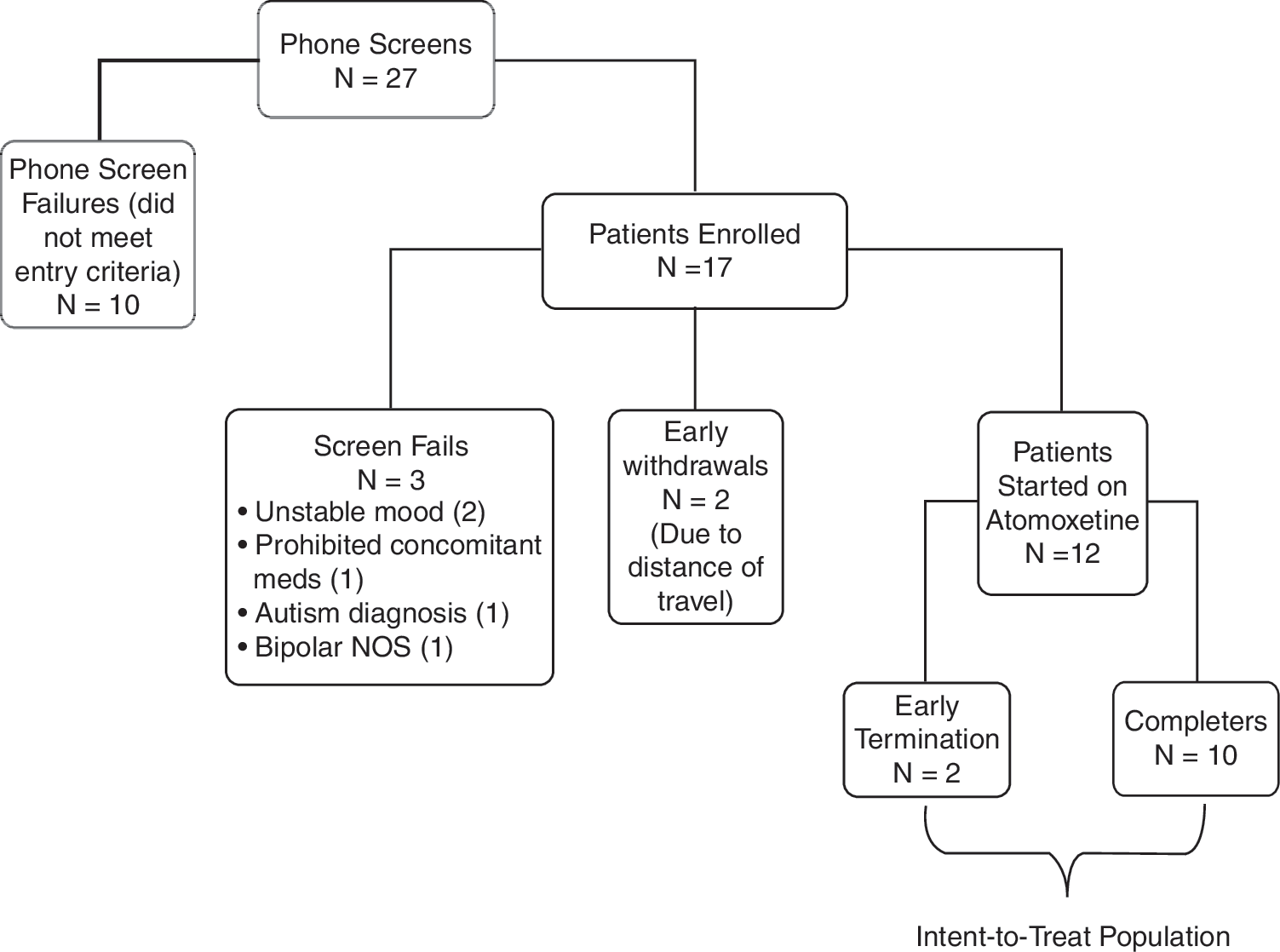
Participant flow chart.
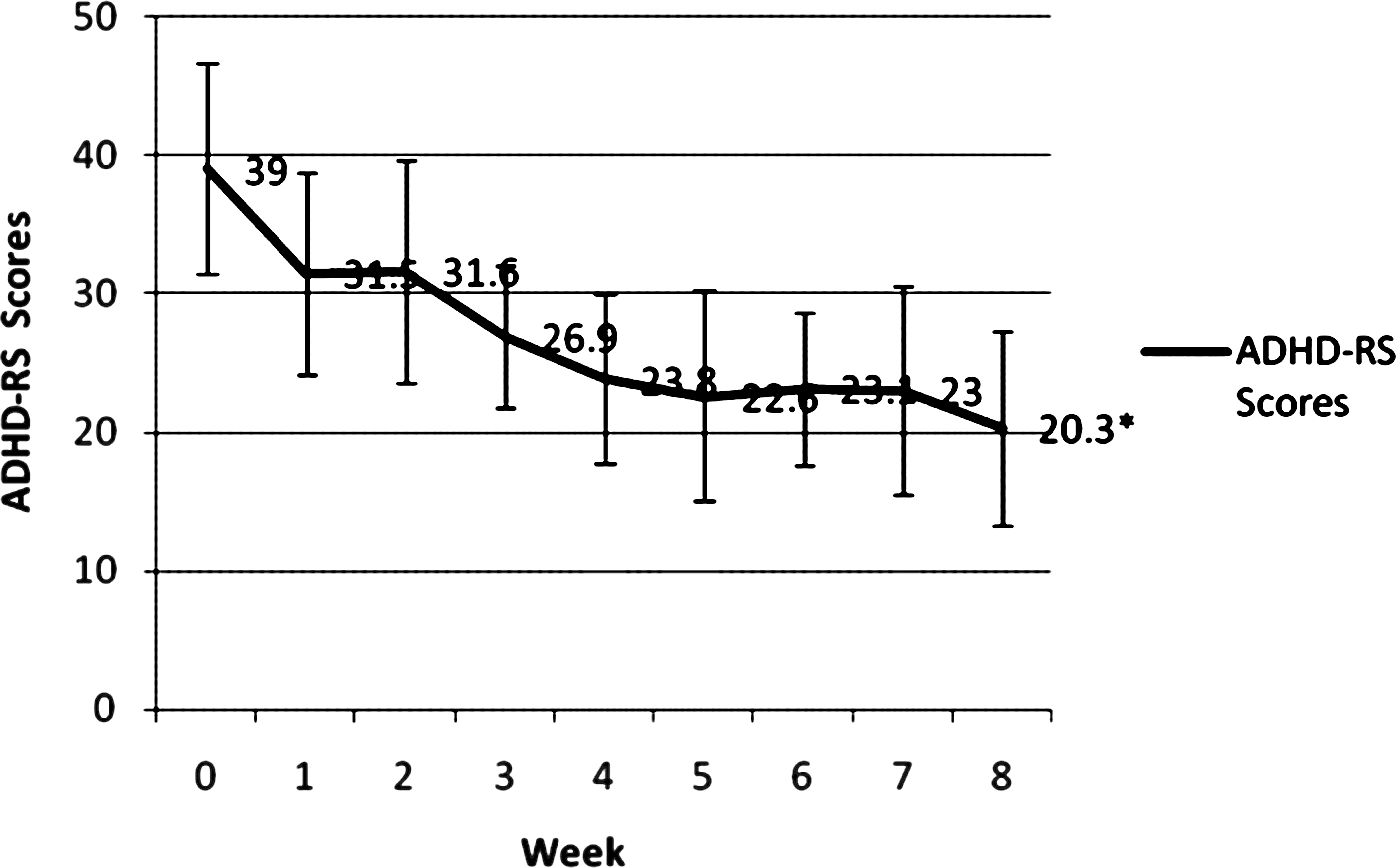
Mean ADHD score by week (data is not LOCF). *p < .0001. ADHD, Attention-deficit/hyperactivity disorder; LOCF, last observation carried forward; ADHD-RS, ADHD Rating Scale.
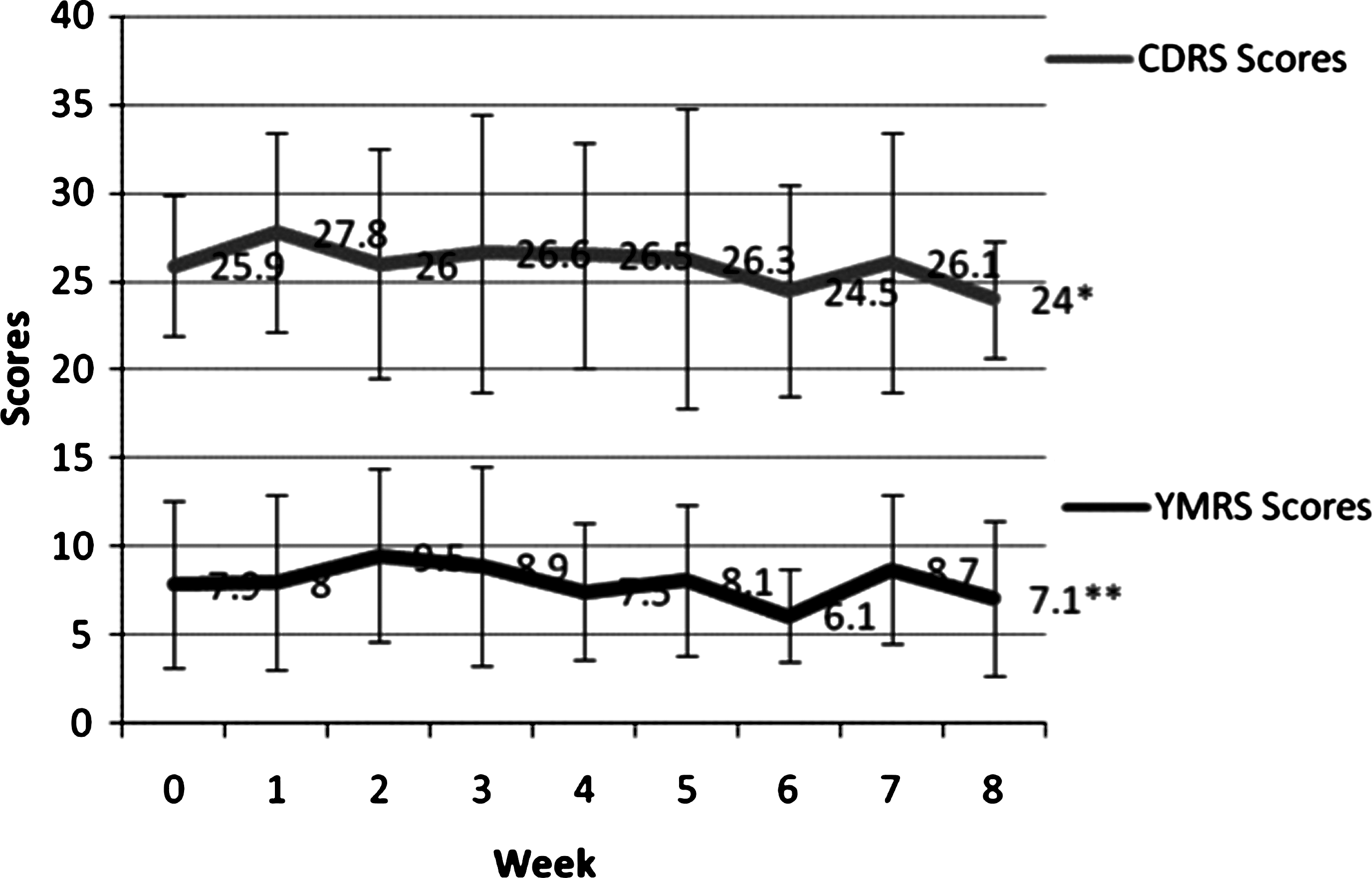
Mean YMRS and CDRS scores by week (data is not LOCF). *p = .71; **p = .72. YMRS = Young Mania Rating Scale; CDRS = Children's Depression Rating Scale; LOCF = last observation carried forward.
Abbreviations: SD = standard deviation; ADHD = attention-deficit/hyperactivity disorder; ODD = opposition defiant disorder; ADHD-RS = ADHD Rating Scale; YMRS = Young Mania Rating Scale; CDRS =Children's Depression Rating Scale; CGI-BP-S = Clinical Global Impressions–Bipolar Disorder–Severity; BMI = body mass index; BP = blood pressure.
Adverse events included tiredness (25%), stomachache (25%), agitation (16%), nausea (8%), dizziness (8%), anticholinergic reaction (8%), and suicidal ideation (8%). The mean weight change was a decrease of 1.4 kg. The mean baseline body mass index (BMI) was 24.63, with a decrease of 0.66 by termination. There were no clinically significant changes in blood pressure, pulse, laboratory values, including liver function tests, or mood stabilizer levels.
Discussion
We found atomoxetine to be efficacious in treating symptoms of ADHD in children and adolescents with BPD taking mood stabilizers and/or antipsychotics, in those subjects who tolerated 8 weeks of treatment. Overall there was a significant decrease in ADHD-RS scores, with high rates of remission (50% by a priori defined criteria). The mean week 8 ADHD-RS score of 20.3 indicates residual presence of some symptoms of ADHD, approximately 1–1.5 SD from population norms. The effect size was 0.73, a moderate effect size very close to that found in a large study of once-a-day atomoxetine versus placebo in children and adolescents with only ADHD (0.71) (Michelson et al. 2002). This finding suggests that children with BPD and ADHD who are already taking a mood stabilizer or antipsychotic may respond similarly to atomoxetine.
Furthermore, overall there was no significant change in mania or depression severity ratings. No subject experienced a manic or mixed episode during study. However, one subject experienced suicidal ideation during week 2 of treatment. This subject had a history of severe suicidal ideation and attempts, but none within the previous 6 months. After cessation of atomoxetine and adjustment of her other medications, the suicidal ideation resolved. Another subject had 2 consecutive weeks of hypomanic symptoms necessitating removal from the study. This subject also had a history of such episodes, but none for the 3 weeks preceding starting atomoxetine. Due to the open-label nature of this study and lack of a placebo comparison group, it is not possible to discern if this worsening of mood symptoms was due to the atomoxetine or the natural course of BPD in these subjects. Of note, suicidal ideation, while uncommon, was at an incidence greater than placebo in a meta-analysis of atomoxetine studies (Bangs et al. 2008). However, it should also be noted that no completed suicides occurred in these studies, and the effects of atomoxetine regarding suicidality in this population of children with BPD is unknown. Nonetheless, atomoxetine does carry a “black box warning” for possible suicidal thoughts, as mandated by the FDA.
Atomoxetine was generally well tolerated physically, with no significant changes in blood pressure or BMI, with a mild mean decrease in weight. This decrease in weight in most cases was welcome, as children in this study were largely overweight, reflecting the typical child with BPD taking mood stabilizers and antipsychotics, most of which may cause weight gain (Correll 2007). The mean weight change over 6–9 weeks in studies of atomoxetine in children with uncomplicated ADHD ranged from −0.4 kg to −0.9 kg (Michelson et al. 2001; Michelson et al. 2002; Spencer et al. 2002). Our subjects may have had slightly more weight loss (−1.4 kg) due to their largely overweight status. The most common adverse effects reported in our study (somnolence, stomach upset) were similar to those also reported in these prior placebo-controlled studies (Michelson et al. 2001; Michelson et al. 2002). Because atomoxetine also carries a “black box warning” for possible liver toxicity, we specifically investigated liver function tests and found no cases of liver function test elevations.
This study was limited by the small number of participants. It should be noted that this study was difficult to recruit for, because participants needed to be “euthymic,” as defined by 3 weeks of relatively low severity of manic and depressive symptoms. Several (27) children were screened and followed, but few (44%) were eligible or able to maintain 3 weeks of euthymia. These children were also not taking ADHD medications during this time, and because symptoms of ADHD overlap with those of mania (Spencer et al. 2001), YMRS scores were consistently fairly high. This difficulty also reflects the frequent chronicity of mood symptoms in children with BPD (Birmaher et al. 2006; Geller et al. 2008). A further obvious limitation was the open-label nature of this study, and, as such, findings may be due to placebo effects. Nonetheless, these findings suggest that in addition to stimulants (Scheffer et al. 2005; Findling et al. 2007), atomoxetine may offer another treatment option for ADHD co-morbid with BPD in children and adolescents. Further placebo-controlled studies with larger samples are needed to clarify the role of atomoxetine in the treatment of ADHD co-morbid with pediatric BPD.
Disclosures
Within the past 2 years, Kiki D. Chang, M.D., has received research grants from AstraZeneca Pharmaceuticals, Eli Lilly and Company, Otsuka America Pharmaceutical, Inc., and GlaxoSmithKline; is on the Speakers Bureau for Abbott Laboratories, AstraZeneca Pharmaceuticals, Bristol-Myers Squibb, and Eli Lilly and Company; is a consultant for Abbott Laboratories, AstraZeneca Pharmaceuticals, GlaxoSmithKline, Eli Lilly and Company, and Shire Pharmaceuticals; and is on the advisory board of Abbott Laboratories and Eli Lilly and Company. Ms. Nayar, Ms. Howe, and Dr. Rana have no conflicts of interest or financial ties to disclose.
Footnotes
This study was funded by a research grant from Eli Lilly and Company.