
Abstract
Objective:
The aim of this study was to evaluate dose–response characteristics in adolescents with attention-deficit/hyperactivity disorder (ADHD) treated with once-daily OROS® methylphenidate (OROS® MPH) during the 4-week, open-label, escalating dose-titration phase of a larger multisite, placebo-controlled trial. Patient factors such as age, height, weight, and baseline symptom severity were evaluated as predictors of selected dose, as was the degree of incremental response with each successive dose escalation.
Methods:
Adolescents 13–18 years of age with ADHD underwent a 4-week, open-label, escalating dose-titration trial to determine the minimal effective dose (18, 36, 54, or 72 mg once daily) of OROS® to be used in a multiphase, placebo-controlled study (NCT00249353). Both final absolute dose and mean weight-adjusted dose were used to assess predictors of response, using a one-way analysis of variance and regression analyses.
Results:
The majority of subjects who did not respond at lower doses achieved response at each escalating dose level. Approximately two-thirds of subjects required a dose of 54 mg or greater to achieve improvement criteria. Minimal effective dose correlated modestly with baseline symptom severity. Age, height, and weight did not correlate with absolute dose and accounted for only a small percentage of variance in weight-based dose. Weight was not a major factor in predicting effective dose; however, using weight-adjusted rather than absolute dose proved slightly superior for modeling of adverse effects.
Conclusions:
Adolescents required, on average, a higher absolute dose but a lower weight-adjusted dose (mg/kg) of OROS® than was previously reported in children. There were few predictors of optimal dose of OROS® other than baseline symptom severity. The increased percentage of adolescent responders at each dose level using this clinically driven approach to titration differs from recent findings from randomized forced dose titration studies in adults with ADHD.
Introduction
Although the beneficial effects of methylphenidate (MPH) have been demonstrated in adolescents with ADHD (Smith et al. 2000; Evans et al. 2001; Stein 2003), doses used for MPH treatment in adolescents have often been based on the multitude of research in school-aged children. However, adolescents with ADHD differ from children in that they are larger and generally display fewer hyperactive-impulsive symptoms (Barkley et al. 1990; Hart et al. 1995; Biederman et al. 2000). Given such differences, dosing recommendations in adolescents should not be based on data from samples comprised predominantly of school-aged children.
Over the past decade, dosing recommendations for stimulants have often followed a fixed rather than a weight-based strategy (Greenhill et al. 2002a; Greenhill et al. 2002b). Children who do not respond at lower dose levels have a higher probability of symptomatic improvement as a function of increasing dose (Rapport et al. 1994). Similar findings have been reported in studies of adults with ADHD (Spencer et al. 1995; Faraone et al. 2004). As adolescents gain weight and stature, it is reasonable to assume that they may require a larger daily dose of medication to achieve or maintain response, but this has not yet been established. Results of studies in both children and adults suggest that higher target daily doses of MPH (i.e., 1.0 mg/kg per day) result in more robust improvement in ADHD symptoms than lower daily doses (i.e., 0.5 mg/kg per day) (Faraone et al. 2004). However, findings from randomized fixed dose trials with OROS® (Medori et al. 2008), d-MPH extended release (Spencer et al. 2007), and mixed amphetamine salts extended release (Weisler et al. 2006) cast doubt on this conclusion because change in ADHD symptom scores was not appreciably different at the different dose levels studied.
OROS® MPH is a long-acting, controlled-release formulation of MPH designed to release MPH gradually with clinical efficacy lasting up to 10–12 h (Pelham et al. 2001; Swanson et al. 2003). The efficacy and safety of OROS® has previously been demonstrated in children at doses up to 54 mg once daily (Swanson et al. 2000; Pelham et al. 2001; Wolraich et al. 2001; Stein 2003; Wilens et al. 2005) and in adults at doses up to 72 mg/day (Medori et al. 2008). OROS® was also demonstrated to be effective and well tolerated at doses up to 72 mg/day in adolescents with ADHD during the placebo-controlled, double-blind phase of this multisite study (Wilens et al. 2006).
To better understand characteristics of dose–response using a clinically driven, escalating approach to titration, and predictors of effective dosing of OROS®in adolescents, a subanalysis was conducted from the adolescent study data set, using doses up to 72 mg/day. The potential moderating effects of age, height, weight, and baseline symptom severity on selected dose were examined.
Methods
Subjects
Adolescents 13–18 years of age with a diagnosis of ADHD based on the Diagnostic and Statistical Manual of Mental Disorders, 4th edition (DSM-IV), criteria (American Psychiatric Association 1994) were recruited for participation in the study (Wilens et al. 2006). Eligible subjects also had to have ADHD confirmed by the Kiddie Schedule for Affective Disorders and Schizophrenia structured diagnostic interview (Kaufman et al. 1997) and a Children's Global Assessment Scale (Shaffer et al. 1983) rating of 41–70 inclusive. The full details of the study design, methods, primary efficacy and safety measures, and results have previously been reported.
Eligible subjects receiving psychosocial treatment, including behavioral modification, were permitted to enroll, provided they did not plan to initiate a new psychosocial program during the study period. Subjects with known hypersensitivity to MPH or a history of nonresponse to MPH were excluded from the study, as were subjects with psychiatric co-morbidity requiring additional or different medication and/or those with a history of glaucoma, seizure disorder, Tourette's syndrome, bipolar disorder, or an eating disorder. Medical exclusion criteria included treatment with theophylline, coumarin, or anticonvulsants; severe gastrointestinal narrowing; or systolic or diastolic blood pressures at the 95th percentile or greater for age, sex, and height at screening.
Informed consent procedures
Caregivers were required to give signed informed consent and each subject was required to give written assent. The study was approved by the institutional review boards of the participating centers or by the California Research Advisory Panel for centers in the state of California, and it was conducted in accordance with the Declaration of Helsinki and its amendments.
Study design
This multiphase trial of OROS® consisted of the following periods: A 1-week washout for patients currently taking medication prior to beginning the study; an open-label, dose-titration phase to select a minimal effective dose of OROS®; a 2-week, double-blind, placebo-controlled, dose-efficacy assessment phase; and an 8-week, open-label, follow-up safety-assessment phase. Baseline symptom measures were performed at the end of the 1-week washout period, during which no ADHD medications were administered. A previously published report focused on results of the randomized efficacy study, which contrasted findings achieved using the minimally effective, individually selected dose of OROS® versus placebo treatment (Wilens et al. 2006). Here we present a fine-grained analysis of data collected during the open-label, dose-titration period, the purpose of which was to identify the individualized dose of OROS® associated with positive clinical response. The response threshold was predefined as a 30% or greater reduction in the baseline ADHD Rating Scale (ADHD-RS) total score and a rating of “good” or “excellent” on the Global Assessment of Effectiveness scale, an instrument that was designed to describe the clinician's overall assessment of treatment efficacy rated on a 4-point scale (i.e., poor, fair, good, excellent) (Wilens et al. 2006). Because the sequential dose-titration approach used in the open-label phase of this study reflects standard-of-care clinical procedures (Greenhill et al. 2002b), the dose–response findings are potentially useful for informing clinical practice.
All subjects initiated therapy at 18 mg/day, and clinical response was measured after 1 week. If response to treatment was inadequate, as per the a priori study definition, the dose was titrated upward (in 18-mg increments) at 1-week intervals for up to 4 weeks, with the maximum dose being 72 mg/day. Subjects concluded the open-label, dose-titration phase when the response criteria had been attained or after treatment with the highest study dose (72 mg). The dosage of OROS® could not be adjusted after subjects reached the designated improvement criteria, even if additional improvement was deemed possible. Thus, we refer to the dose selected in this trial as being the minimally effective dose.
Statistical analyses
Post hoc analyses examined the potential moderating effects of various patient characteristics on dose–response characteristics and tolerability of OROS®. Baseline demographics, efficacy, and safety data were analyzed using chi-squared or Fisher exact tests for categorical variables and one-way analysis of variance for continuous variables.
For dimensional measures, analysis of covariance was used, with study-site and treatment group as factors and baseline score as the covariate. Statistical significance was set at a p value of 0.05 and all tests of hypotheses were two-sided. Characteristics such as patient age, height, weight, and baseline symptom severity were examined as potential moderators of dose. To contrast the utility of fixed versus weight-based dosing strategies, the sample was subdivided into four relatively equal weight-based dose groups based upon the selected minimally effective absolute dose (≤0.61 mg/kg, 0.62–0.83 mg/kg, 0.83–1.10 mg/kg, and >1.10 mg/kg). Predictors of dose–response were evaluated in these weight-based dose quartiles using multiple regression analyses. For variables for which there were significant differences across the four weight-based dose levels, post hoc tests using the Tukey studentized range test were used to identify significant group differences.
Results
Subjects
A total of 220 subjects entered the open-label, dose-titration phase and initiated treatment with OROS® at a dose of 18 mg once daily. The subjects were primarily medication naïve; only 24 indicated they had previously received medication for ADHD (mean exposure = 3.4 years). Subject disposition for this phase of the study is shown in Fig. 1. At the conclusion of the dose-titration phase, 182 (83%) of the original 220 subjects had their dose successfully titrated to an effective once-daily dose of OROS® (Fig. 2). The dose–response relationship was linear; following each dose escalation, an incremental number of nonresponders achieved the predetermined threshold response.

Subject disposition for the open-label, dose-titration phase.
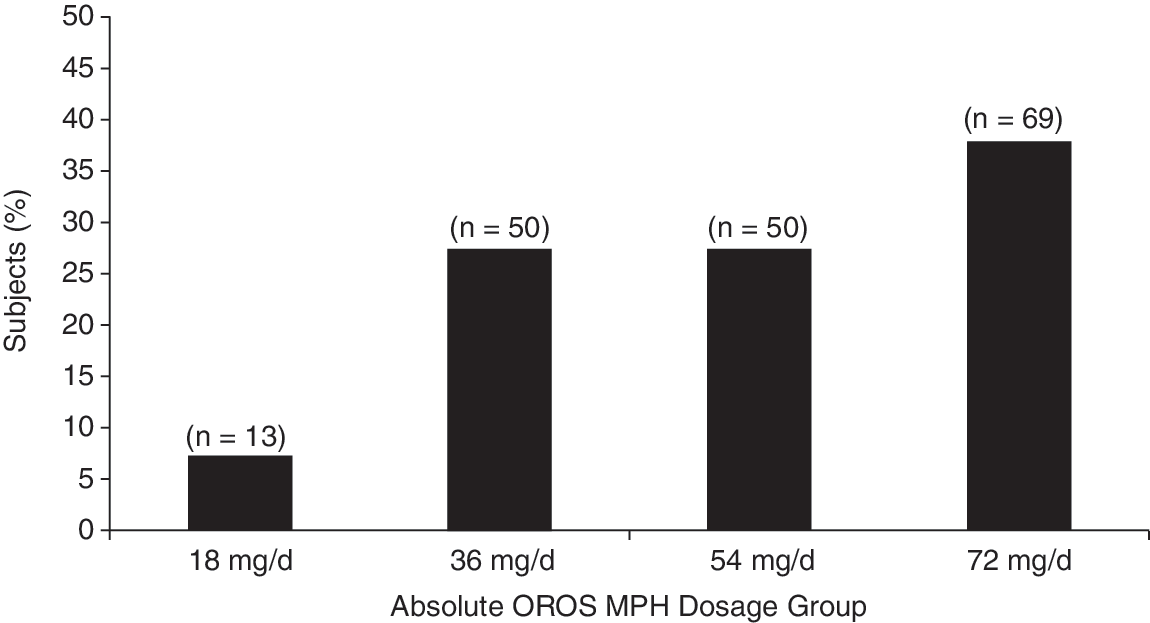
Percentage of adolescent subjects per titrated absolute OROS® dosage group (n = 182). This figure lists 50 subjects (instead of 49) in the 54 mg group and 69 subjects (instead of 70) in the 72 mg group because 1 subject who successfully titrated to 72 mg was randomized to 54 mg. This patient started taking 72 mg in the titration phase but experienced anxiety at this dose level; therefore, the subject was enrolled into the double-blind phase and randomized to the 54 mg group. OROS® = OROS® methylphenidate.
Absolute dose groups
Mean values for weight, height, and age for subjects in the four OROS® absolute-dose groups (i.e., 18 mg/day, 36 mg/day, 54 mg/day, and 72 mg/day) at the conclusion of the open-label titration are listed in Table 1. These values did not differ across dose groups. Pearson correlation coefficients indicated that there were no significant relationships between dose and subject weight (0.13; p = 0.075), height (0.07; p = 0.378), or age (0.11; p = 0.146), although one could interpret the weight finding as representing a trend. As expected, there was a linear increase in the weight-adjusted dose across the four dose groups: 0.28 mg/kg (18-mg group), 0.63 mg/kg (36-mg group), 0.84 mg/kg (54-mg group), and 1.12 mg/kg (72-mg group).
This table lists 50 subjects (instead of 49) in the 54 mg group and 69 subjects (instead of 70) in the 72 mg group because 1 subject who successfully titrated to 72 mg was randomized to 54 mg. This patient started taking 72 mg in the titration phase but experienced anxiety at this dose level; therefore, the subject was enrolled into the double-blind phase and randomized to the 54 mg group.
Abbreviations: ADHD = Attention-deficit/hyperactivity disorder; bpm = beats per minute; SD = standard deviation.
Weight-based dose quartile groups
To further analyze the potential contribution of size to the final titrated dose, subjects were stratified into four weight-based dose groups, each representing approximately 25% of the study population: ≤0.61 mg/kg (n = 47), 0.62–0.83 mg/kg (n = 48), 0.83–1.10 mg/kg (n = 43), and >1.10 mg/kg (n = 44) (Table 2). Two of the dose-quartile groups were below and one group was above the mean dose in the study. The third quartile (0.83–1.10 mg/kg) roughly brackets the clinical dose of 1 mg/kg per day previously used successfully in treating ADHD in adults (Spencer et al. 1995).
Abbreviations: ADHD = Attention-deficit/hyperactivity disorder; bpm = beats per minute; SD = standard deviation.
Significant dose–response differences were found for several variables in the weight-based but not the absolute dose analyses, including age, height, systolic blood pressure (significant in both analyses but more clearly shown using the weight-based groupings), baseline investigator-rated ADHD-RS total score, and parent- and investigator-rated hyperactivity subscale scores. The parent-rated ADHD-RS total score was significant in both the weight-based and absolute dose group analyses. The only variable that differed in the absolute but not the weight-based dose group analysis was baseline inattention (both parent- and investigator-rated) (Tables 1 and 2).
Weight-based versus absolute dosing
Although weight itself was not a strong predictor of dose, both visual examination of the patterns of group differences and application of post hoc significance testing suggested that dose effects are modeled somewhat better using weight-based rather than absolute dosing. Weight was somewhat lower in the two higher weight-based dose groups, demonstrating that when absolute dosing is applied, younger and lighter adolescents end up receiving higher mg/kg doses. The linear trend for increased baseline ADHD symptom severity as mg/kg dose increases can be seen in both the absolute dose and weight-based dose analyses. However, it is a bit more sensitively modeled using the weight-based assessment strategy, with increased baseline severity being a predictor of higher dose. This increase in symptom severity was not specifically associated with either inattentive or hyperactive symptoms, although the dose–response relationship for hyperactive symptoms was more clearly illustrated using the weight-based schedule. Although the data were not analyzed categorically as a function of ADHD subtype, the latter finding might lead us to conclude that cases of ADHD combined subtype were present in a higher proportion in the highest weight-based dose quartile.
Regression analysis
A regression analysis was performed using stepwise selection to determine predictors of dose–response among the following factors: Age, weight, height, baseline investigator-rated ADHD-RS, and baseline parent-rated ADHD-RS. For both weight-based and absolute dosing, the parent-rated ADHD-RS was a significant factor (p ≤ 0.0029) in determining dose, although the parameter estimate indicates that this was a small effect. As in the group analyses, weight was not a significant factor in explaining absolute dose. Although parent-rated ADHD-RS scores showed significance in the model and investigator-rated scores did not, these measures are known to be highly intercorrelated (Goldman et al. 1998). When the parent-rated measure was removed from the model, investigator-rated scores were shown to be significant predictors of both weight-based and absolute dose.
Adverse events
There was no relationship between incidence or severity of spontaneously reported adverse events and increasing absolute dose. One or more drug-related adverse events were reported by 126 (57.3%) subjects who were treated with OROS® during this open-label, dose-titration study, with the incidence of drug-related adverse events being similar across absolute dose groups (Table 3). The percentage of adverse events reported ranged from 24.4% in the 36-mg group to 35% in the 18-mg group. The large majority of the reported adverse events were mild or moderate in intensity in all dose groups. The dose–response relationship was linear; following each dose escalation, an incremental number of nonresponders, who tolerated the preceding dose adequately, achieved the predetermined threshold response.
OROS MPH dosage was titrated upward to an effective dose during the study, so subjects may be counted in more than one dose group. Adverse events are reported at the dose at onset of the adverse event. Drug-related adverse events include adverse events with relation to drug of “certain,” “probably/likely,” “possible,” or “unknown.”
Abbreviation: MPH = Methylphenidate.
Discussion
The purpose of this paper was to evaluate the predictors of effective dose for OROS® in adolescents 13–18 years of age with ADHD. Additionally, we report on the number of incremental responders using a sequential, escalating approach to dose titration that mimics dosing in clinical practice. The findings demonstrate that the majority of adolescents who do not respond to any given dose, but who tolerate the medication, are able to achieve response when their dose is raised. This was true for every dosing increment (i.e., 18–36 mg; 36–54 mg; 54–72 mg). The only significant predictor of effective dose was baseline ADHD symptom severity, although the relationship was only modest. Age, height, and weight did not predict final dose. Nevertheless, there was a slight advantage for using a weight-based approach for modeling adverse effects. This is potentially noteworthy, given that the experience of adverse events is likely related to tolerability, and therefore compliance. We conclude that in adolescents it may be important to be mindful of weight-based dose, even though titration will often be conducted using an absolute-dose approach.
In clinical practice, dose titration of stimulants often proceeds in a gradual, stepwise manner, from lower to higher doses. According to the American Academy of Child and Adolescent Psychiatry practice guidelines, the minimum effective dose of MPH should be used to initiate therapy in children and adolescents with ADHD, with the dose increased in weekly increments of 5 or 10 mg per dose if symptom control is not achieved (Greenhill et al. 2002b). Partial response is not uncommon at lower doses, and failure to systematically test higher doses may decrease the likelihood of achieving optimal responses (Greenhill et al. 1996). Our findings—using an open, escalating-dose titration—support this contention, because a large number of adolescents who did not respond at lower doses responded as the dose was raised, at every dose-escalation point. This increase in response was generally achieved without increased risk of adverse effects. Interestingly, although some studies of children with ADHD have found a dose–response relationship with MPH treatment (Greenhill et al. 2001) similar to what we report here, this has not always been the case, particularly for specific ADHD subgroups. For example, Stein and colleagues (Stein et al. 2003) reported a linear dose–response relationship for OROS®, favoring higher doses in children with the Combined subtype, but not children with Inattentive subtype, in whom the greatest relative benefit was observed at lower doses. Our findings in adolescents that baseline symptom severity was positively correlated with effective dose are consistent with data in children in the Multisite Multimodal Treatment Study of Children with Attention-Deficit/Hyperactivity Disorder (MTA) study, which found that lower baseline severity was associated with increased likelihood for achieving an optimal response (Owens et al. 2003).
The majority of adolescents in this study (approximately two-thirds of the sample) required dosing to either 54 or 72 mg of OROS® to reach the prespecified response threshold. The absolute dose associated with response in this sample was higher than was previously demonstrated in 6- to 12-year-old children (53 mg vs. 34.3 mg) (Wolraich et al. 2001). Although the mean absolute dose required for response in this study was 54% greater than in the child study, the mean weight-adjusted dose was actually lower in adolescents (0.84 ± 0.33 mg/kg) compared with children (1.1 ± 0.5 mg/kg) (Wolraich et al. 2001). Understanding the similarities and differences between absolute and weight-based dosing may be helpful to clinicians who are less familiar with dosing strategies in older populations.
Findings from this study indicating an association of increased baseline severity and higher dose in adolescents parallel those in children in the MTA study. However, this study included any ADHD subtype and the MTA study only included children with Combined subtype (Greenhill et al. 2001). The current study therefore extends the MTA finding to adolescents and to a subject group that includes all ADHD subtypes. Stein and colleagues (2003) also found an association between symptomatology and dose, but in that study, successful treatment at a lower dose was restricted to the inattentive subtype. Of course, the Inattentive subtype is generally characterized by lower overall ADHD-RS scores, and in that sense, the findings from Stein et al. (2003) and the current study are compatible.
The need for a higher absolute dose of OROS® to treat adolescents with ADHD in this study is consistent with findings from other studies of MPH in adolescents (Smith et al. 2000; Evans et al. 2001), but it is also at variance with some published findings. For example, Evans and associates (2001) reported positive effects of immediate-release MPH on classroom behavior and academic performance of adolescents at doses of 10, 20, and 30 mg; however, the degree of improvement on active medication versus placebo was greater for the 10-mg dose and therapeutic gain leveled out at 20 and 30 mg. In contrast, and similar to the findings of this study, Findling and colleagues (2001) found that adolescents required a smaller weight-adjusted dose of stimulant medication than children. They concluded, as we do, that adolescents with ADHD do not necessarily require a larger dose of stimulant medication than children, but that depends on how one calculates dose.
Of note, our findings using an escalating dose-titration approach, in which higher doses are offered to nonresponders at lower doses who tolerated the medication, differ from those in studies of adults using a forced dose-titration methodology. For example, in a forced dose-titration study of OROS® in adults, the mean change from baseline to end of study on the Conners' Adult ADHD Rating Scale: Observer form (CAARS:O-SV) total score (last observation carried forward [LOCF]) was greater (p < 0.015) in subjects randomized to receive either 18 mg, 36 mg, or 72 mg of OROS® compared with placebo, but the degree of symptom change in the three OROS® treatment groups was not significantly different (Medori et al. 2008). Similarly, Spencer and colleagues (Spencer et al. 2007) found no difference in symptomatic improvement in adults on the ADHD-RS between randomized dose groups receiving 20 mg, 30 mg, or 40 mg of d-MPH extended release, although there was a higher number of responders (defined as those who were much or very much improved on the CGI-Improvement scale) in the 40-mg group than the 20-mg group. A study that randomized subjects to placebo, and 20 mg, 40 mg, or 60 mg of mixed amphetamine salts extended release produced similar results (Weisler et al. 2006). Of note, the latter two studies informed the FDA decisions to establish 20 mg as the upper dose level of d-MPH extended release and mixed amphetamine salts extended release in adults, despite the fact that the approved upper dose levels in children are higher. These findings have raised questions as to whether it is necessary to use higher absolute doses of stimulants as people gain in age and size. However, the findings of the current study indicate that a clinically driven approach to titration in adolescents, assessing response and tolerability of lower doses before treating with higher doses, and restricting use of higher dose treatment to those patients most likely to tolerate and respond, does indeed produce incremental improvement as the dose is increased.
The relatively higher absolute doses of stimulants used in adolescents appear to be well tolerated. The incidence of adverse events in this study did not increase significantly as a function of increasing absolute dose. Nevertheless, considering dose in a weight-adjusted format proved to be slightly more sensitive for modeling potential adverse effects. Finally, our findings indicate the dosing parameters of OROS® needed to treat most adolescents with ADHD. Given the mean daily dose of 0.84 mg/kg in this study, a target dose of approximately 1.0 mg/kg per day, reached through sequential stepwise titration, should be sufficient for most adolescents, as it is in other age groups.
These data must be considered in light of several limitations in design. The dosing determinations in this study were made during open-label titration, not under double-blind conditions. Nevertheless, an open, dose-escalating titration methodology most clearly reflects clinical practice, although the current study limited titration to 4 weeks. Furthermore, although this study tested a higher dose (i.e., 72 mg) than was used in previous studies of OROS®, it did not attempt to define the maximum effective tolerated dose. It is possible that the incremental improvement observed when increasing dose in nonresponders from 36 mg to 54 mg, and 54 to 72 mg, might also have been observed if the dose were further increased from 72 to 90 mg in nonresponders. However, if a subject did not achieve response at 72 mg in this study, he or she was discontinued. In addition, titrating to a more substantive level of improvement (i.e., more than a 30% decrease in symptoms) would likely have increased the selected dose for many subjects. Of note, the threshold for improvement used in this study was based on the standard used in studies of stimulants in adults (Taylor et al. 2000; Spencer et al. 1995; Spencer et al. 2001; Taylor et al. 2001; Adler et al. 2009), and exceeded the threshold of 25% reduction in ADHD symptoms more typically used in studies with children at that time (e.g., Faries et al. 2001; Michelson et al. 2002). Nevertheless, use of a minimally effective dose as the threshold for improvement in this study likely underestimated the dose required to achieve symptomatic remission, which is currently considered the goal of therapy (Steele 2006). Finally, although we report dose–response characteristics here, this was not a pure dose-titration study. Subjects could not revert to lower doses once they had been titrated to the next higher dose—even if there were problems in tolerability at the higher dose. For instance, if a subject reported a new adverse event at 54 mg, the dose could not be titrated downward to 36 mg because it had already been determined that the lower dose did not provide sufficient symptom response to meet the necessary improvement criteria. This, obviously, differs from the approach used in clinical practice.
Conclusions
Adolescents may be effectively and safely treated with once-daily doses up to 72 mg of OROS®, with the majority requiring doses of 54 or 72 mg to achieve an adequate response. Adolescents are likely to require higher absolute doses than younger children, but their weight-adjusted dose is not higher than it is in children. Although weight, height, and age do not appear to be substantive predictors of final OROS® dose, ADHD symptom severity, particularly hyperactive-impulsive symptomatology, does appear to predict dose. The latter finding is important because severity has also been demonstrated to be a predictor of persistence. Further study is required to evaluate other possible dose–response relationships associated with OROS® treatment in adolescents with ADHD, including the degree to which ADHD subtype may moderate the best-tolerated dose. Study results highlight the importance of considering baseline severity and weight-based dose in attempting to optimize treatment with OROS®.
Footnotes
Disclosures
At the time of the trial, Kimberly M. Cooper was an employee of McNeil Pediatrics. She is currently employed by Johnson & Johnson Pharmaceutical Research & Development. Mark A. Stein receives research support from Novartis, Organon, Ortho-McNeil-Janssen Scientific Affairs, LLC, McNeil, and Lilly. He is also on the Speakers Bureau for Novartis, and is an advisor to Shire and Novartis. Jeffrey Newcorn is a recipient of grants for research support from Eli Lilly, Ortho-McNeil-Janssen Scientific Affairs, LLC, and Shire. He is also a consultant and/or advisor for Eli Lilly, Novartis, Ortho-McNeil-Janssen Scientific Affairs, LLC, Shire, Schering-Plough, Astra-Zeneca, and BioBehavioral Diagnostics.
Acknowledgment
The authors would like to acknowledge the editorial assistance of Wynne Dillon from JK Associates, Inc. in the preparation of this manuscript.
Ortho-McNeil Janssen Scientific Affairs, LLC, Raritan, New Jersey, funded the editorial, writing, and technical assistance provided by Wynne Dillon from JK Associates, Inc. in the completion of this manuscript. McNeil Consumer & Specialty Pharmaceuticals, a Division of McNeil-PPC, Inc., sponsored the study.