
Abstract
Aim:
To report the long-term efficacy of aripiprazole in the treatment of irritability in children and adolescents (ages 6–17 years) with autistic disorder.
Methods:
This was a 52-week, open-label, flexible-dose (2–15 mg/day) study of aripiprazole for the treatment of children and adolescents with irritability associated with autistic disorder. Eligible subjects were enrolled from two 8-week randomized trials or were enrolled as de novo subjects. “Prior aripiprazole” subjects had received treatment with aripiprazole for 8 weeks before entering this study. Evaluation of efficacy, a secondary objective after evaluation of safety and tolerability in this study, was conducted using the caregiver-rated Aberrant Behavior Checklist–Irritability subscale and the clinician-rated Clinical Global Impression–Improvement score.
Results:
Three hundred thirty subjects received treatment (de novo, n = 86; prior aripiprazole, n = 174; prior placebo, n = 70) and 199 subjects (60.3%) completed 52 weeks of treatment. At their last study visit, 38.2% of subjects were receiving concomitant central nervous system medications (commonly antidepressants, 13.4%; psychostimulants, 11.5%; antiepileptics, 5.9%). At week 52 (observed cases data set), the mean change from baseline in Aberrant Behavior Checklist Irritability subscale scores was −8.0 in de novo subjects and −6.1 in prior placebo subjects; prior aripiprazole subjects maintained symptom improvement that was achieved with treatment in the prior study. At endpoint, the majority of subjects had a Clinical Global Impressions–Improvement score of 2 (much improved) or 1 (very much improved).
Conclusion:
Aripiprazole reduced symptoms of irritability associated with autistic disorder in pediatric subjects ages 6–17 years who were studied for up to 1 year.
Introduction
Although there are no approved pharmacologic treatments that target the core deficits of autistic disorder, associated secondary symptoms such as irritability may be ameliorated by a combination of behavioral and pharmacologic approaches, including the use of atypical antipsychotics (Myers and Johnson 2007). Risperidone and aripiprazole are approved by the U.S. Food and Drug Administration for the treatment of pediatric patients with irritability associated with autistic disorder, including symptoms of aggression toward others, self-injuriousness, temper tantrums, and quickly changing moods (Aripiprazole 2009; Risperidone 2009).
Recently, aripiprazole was shown to be efficacious and generally safe and well tolerated for the treatment of children and adolescents (ages 6–17 years) with irritability associated with autistic disorder in two 8-week, multicenter, randomized, double-blind, placebo-controlled studies (Marcus et al. 2009; Owen et al. 2009). These studies were similar in design; one was a fixed-dose study in which subjects were randomized to receive aripiprazole (5, 10, or 15 mg/day) or placebo (Marcus et al. 2009), and the other was a flexible-dose study in which subjects were randomized to receive aripiprazole (2–15 mg/day, titrated to clinical effect, with dose options of 5, 10, or 15 mg/day) or placebo (Owen et al. 2009). In the fixed-dose study, all aripiprazole doses produced significantly greater improvement than placebo in mean Aberrant Behavior Checklist–Irritability (ABC-I) subscale scores (5 mg/day, −12.4; 10 mg/day, −13.2; 15 mg/day, −14.4; vs. placebo, −8.4; all p < 0.05) at week 8; in the flexible-dose study, aripiprazole also produced significantly greater improvement than placebo (−12.9 vs. −5.0; p < 0.001).
Although these 8-week studies demonstrated the short-term efficacy, safety, and tolerability of aripiprazole, there remains a need for longer-term data to inform clinical decision making. As such, a 52-week, open-label, multicenter study was conducted to evaluate the long-term safety and tolerability of flexibly dosed aripiprazole for the treatment of irritability associated with autistic disorder. The safety and tolerability findings, which were the primary objective of this study, are reported in detail elsewhere (Marcus et al. In press). Here we report the efficacy findings from this study, which were included as secondary objectives.
Methods
Study design and subjects
This was an open-label, multicenter, 52-week study conducted at 53 sites in the United States between September 2006 and June 2009 in accordance with the Declaration of Helsinki and in accordance with Good Clinical Practice as defined by the International Council on Harmonization. The Institutional Review Board at each site approved the protocol. All parents/guardians provided written informed consent to participate, and subjects provided written, informed assent when possible.
Subjects were children and adolescents, aged 6–17 years, with a diagnosis of autistic disorder and with behavioral problems such as irritability, agitation, self-injurious behavior, or a combination of these symptoms. Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision, criteria for autistic disorder were confirmed using the Autism Diagnostic Interview–Revised (ADI-R) (Lord et al. 1994), which was administered by an experienced interviewer who had been previously trained and approved as “Research Reliable” on the ADI-R or who had been approved after successful completion of a 2-day rater training course conducted by an ADI-R-certified trainer.
Exclusion criteria included a current diagnosis of bipolar disorder, psychosis, schizophrenia, or major depression, Fragile-X Syndrome, or diagnosis of another disorder on the autism spectrum (pervasive developmental disorder–not otherwise specified, Asperger's disorder, Rett's disorder, or childhood disintegrative disorder).
This study enrolled three groups of subjects: (a) subjects who were previously randomized to placebo in one of two 8-week multicenter, randomized, double-blind, placebo-controlled trials (prior placebo subjects); (b) subjects who were previously randomized to aripiprazole in one of the 8-week trials (prior aripiprazole subjects); and (c) de novo subjects who had not been enrolled in these previous studies.
Prior aripiprazole and prior placebo subjects
Subjects entering open-label treatment from the previously completed trials had all met inclusion criteria specified in the original trials. All subjects were required to have a Clinical Global Impressions–Severity (CGI-S) score ≥ 4 and a caregiver-rated ABC (Aman et al. 1985a, 1985b) Irritability subscale score ≥ 18 at baseline. CGI-S ratings were focused on the severity of irritability (e.g., tantrums, aggression, self injury, and quickly changing moods) associated with autistic disorder, as this was the target symptom for treatment. The ABC Irritability subscale is a 15-item subscale that includes items describing symptoms of irritability, such as temper tantrums, aggression, mood changes, and self-injury. Subjects who completed either previous double-blind study were eligible if continuation of treatment was warranted, as judged by the investigator, and they had no significant protocol violations or clinically relevant adverse events that would preclude their inclusion in the study.
De novo subjects
De novo subjects who had not been enrolled in one of these placebo-controlled studies were also permitted to enroll in this open-label study. Although de novo subjects had to demonstrate current behavioral problems or a history of behavioral problems that were currently being treated with psychotropic medication, there was no requirement of a minimum ABC-I or CGI-S score. Study sites were not permitted to enroll de novo subjects in this study until they had completed enrollment for the randomized, controlled studies, unless approved by the study sponsor. De novo subjects with a history of serious behavioral problems who were being treated with aripiprazole and were doing well were eligible for enrollment if, in the investigator's clinical judgment, they would benefit from study participation. For these subjects (n = 6) prior aripiprazole was discontinued at least 9 days (usually longer) before baseline assessment and entry into the current trial.
Study treatments
Aripiprazole treatment
All subjects received open-label treatment with flexibly dosed (2–15 mg/day) aripiprazole (oral tablet) taken once daily at the same time each day without regard to meals. Aripiprazole was initiated at 2 mg/day, with a target dose of 5, 10, or 15 mg/day (maximum dose, 15 mg/day). All subjects, both de novo and rollover, who participated in the study started therapy with aripiprazole at 2 mg/day. All rollover subjects had study medication re-titrated starting at 2 mg/day at entry into open-label treatment, as both subjects and clinicians were blinded to treatment assignment in the previous double-blind study. Rollover subjects took their first dose of study medication during open-label treatment (day 1) on the day after their week 8 visit of the antecedent double-blind studies. All dose increases, if deemed appropriate by the investigator based on the patient's clinical response and medication tolerability, were made no more frequently than every 4 days and were incremental from the current dose level to the next (dose levels were 2, 5, 10, or 15 mg/day). Dose adjustments could be made at any time based on efficacy and tolerability at the current dose.
Prohibited concomitant therapies
Clonidine, guanfacine, guanabenz, carbamazapine, oxcarbazepine, and antipsychotics other than aripiprazole, that is, medications with the greatest potential to impact the assessment of the effects of aripiprazole for the treatment of irritability associated with autistic disorder, were expressly prohibited during open-label treatment. De novo subjects receiving these medications were washed out at least 4 days before the interim screening visit. Subjects receiving long-acting (depot) neuroleptics were discontinued for at least 1 full cycle plus 1 week before interim screening visit. Prior aripiprazole and prior placebo subjects underwent medication washout of these agents before participating in the previously conducted trials.
Permitted concomitant therapies
Subjects were permitted to receive concomitant medications for the treatment of comorbid psychiatric disorders. As such, psychostimulants, antidepressants, anxiolytics, and mood stabilizers were permitted during open-label treatment unless contraindicated or likely to result in an adverse drug interaction with aripiprazole. Although these concomitant psychotropic medications were prohibited or restricted during the previous placebo-controlled studies, a wider spectrum of concomitant treatments was permitted in this open-label study to improve patient retention and more accurately reflect the actual medication-use patterns in this population.
In this open-label study, and the previous double-blind studies, subjects were permitted to receive antianxiety medication (such as benzodiazepines) and sleep aids (limited to diphenhydramine, non-benzodiazepine hypnotics such as zolpidem or eszopiclone or melatonin) at the investigator's discretion. Diphenhydramine could also be given for serious behavior problems (up to 50 mg/day), and psychotropic medication was permitted for the acute treatment of unforeseen medical events. Anticholinergic therapy (benztropine) or propranolol was permitted for the treatment of extrapyramidal symptoms, although administration of movement rating scales was not to occur if these medications had been received in the last 12 hours.
Nonpharmacologic therapy (e.g., psychotherapy and behavior modification) was permitted provided it was stable before screening and consistent throughout the study.
Assessments and statistical analyses
Evaluating the long-term efficacy of flexibly dosed aripiprazole for the treatment of irritability associated with autistic disorder was the secondary objective of this study. Efficacy was evaluated using the caregiver-rated ABC, which has five subscales designated as Irritability (15 items), Lethargy/Social Withdrawal (16 items), Stereotypic Behavior (7 items), Hyperactivity (16 items), and Inappropriate Speech (4 items). Each individual item on the ABC is scored on a Likert scale from 0 (not at all a problem) to 3 (the problem is severe in degree).
Participants were also assessed on the clinician-rated CGI-Improvement (CGI-I) score and CGI-S score (Guy 1976) and the clinician-rated Children's Yale–Brown Obsessive Compulsive Scale (CY-BOCS) (Scahill et al. 1997) (compulsion scale only).
Quality of life assessments included the caregiver-rated Pediatric Quality of Life Inventory™ (PedsQL™) (Varni et al. 2001, 2002), which rates health-related quality of life, and the Caregiver Strain Questionnaire (CGSQ) (Brannan et al. 1997), a 21-item self-report instrument assessing the impact that caring for children and adolescents with serious emotional, mental, and behavioral disturbances has on the family over the preceding 3 months. The PedsQL-combined scales total score was used in this study; this was calculated as the sum on the PedsQL emotional (5 items), social functioning (5 items), and cognitive functioning (6 items from the PedsQL Multidimensional Fatigue scale) subscale items divided by the number of items answered. Scores are presented on a 0–100 scale, where higher scores indicate better health-related quality of life. The CGSQ Global score is presented as the sum of the three subscales, which were calculated as the averages of the corresponding individual items. CGSQ Global scores are on a 0–15 scale, where higher scores indicate a higher degree of caregiver strain.
Subjects had study visits at the end of weeks 1, 2, and 4 (±2 days from the baseline visit) and at the end of weeks 8, 14, 20, 26, 34, 42, and 52 (±7 days from the end of baseline visit). CGI-S and CGI-I ratings were obtained at all study visits; ABC-I was evaluated at week 4 and all visits thereafter. All other efficacy evaluations were assessed at baseline and endpoint.
All analyses were conducted using the efficacy sample, which included all patients who received at least one dose of study medication during open-label treatment and who had at least one postbaseline efficacy evaluation and corresponding baseline value (if applicable).
Three subject groups based on previous treatment were defined for the purpose of the analyses presented herein: (a) prior aripiprazole subjects, (b) prior placebo subjects, and (c) de novo subjects. For each subgroup, descriptive statistics were calculated for the mean change from baseline for each of the efficacy evaluations, or for mean score in the case of CGI-I. For the purpose of analysis, all baseline values were derived from week 0 (open-label treatment baseline visit) for de novo subjects or the last evaluation during double-blind treatment for prior aripiprazole/placebo subjects. The exception to this was CGI-I scores for prior aripiprazole/placebo subjects in which improvement in symptoms was rated relative to symptom baseline before receiving dosing in the double-blind treatment phase of the antecedent study.
As this was an open-label study, with no placebo or active control group, no formal statistical testing was undertaken; all results are reported as descriptive statistics. Summary statistics are presented, including mean and standard deviation for continuous variables, and frequency and percent frequency for categorical variables. Of note, prior aripiprazole subjects' baseline measurements were taken at the last evaluation during double-blind treatment after having received aripiprazole for 8 weeks. As such, mean changes in efficacy scores in this group were expected to be small over the course of long-term treatment and would merely indicate that previous symptom control was maintained.
Results
Subject disposition and demographics
In total, 330 subjects entered open-label treatment in this study. Ninety-five percent (n = 70/74) of placebo subjects and 97% (n = 174/179) of aripiprazole subjects completing the short-term study chose to enter open-label treatment, giving a total of 244 subjects who had previously participated in either of the two 8-week studies. Eighty-six de novo subjects entered the open-label treatment phase; an additional 23 were screened but did not meet entrance criteria. Six de novo subjects had received aripiprazole treatment before study enrollment. The duration of aripiprazole ranged from 24 to 317 days in three subjects; in the remaining three subjects the exact duration of prior aripiprazole treatment was unknown. Prior doses received were aripiprazole 2 mg/day (n = 2), 5 mg/day (n = 2), and 10 mg/day (n = 1). One subject received aripiprazole 10 mg/day with a dose reduction to aripiprazole 5 mg/day. All six subjects discontinued aripiprazole at least 9 days, usually longer, before baseline assessments and treatment.
Subject disposition during the 52-week, open-label, multicenter study is shown in Table 1. The 52-week, open-label treatment period was completed by 60.3% of subjects. Completion rates were broadly similar among subject subgroups. The most common reason for discontinuation was adverse events (10.6% of all subjects); discontinuation due to adverse events was lower in prior aripiprazole subjects (8.6%) than in prior placebo (15.7%) or de novo subjects (10.5%). In all, 6.1% of all subjects discontinued due to lack of efficacy. Additional reasons for discontinuation, generally unrelated to study medication, included loss to follow-up (9.4% of all subjects), withdrawal of consent (8.2%), inadequate compliance (1.5%), administrative reasons (1.2%), and other (2.9%). A total of 330 subjects received at least one dose of study medication, of whom 322 (97.6%) had at least one postbaseline efficacy assessment; these were included in the efficacy sample.
Subject baseline demographic characteristics have been presented previously (Marcus et al. In press) and were similar among subgroups. The mean age for all subjects was 9.6 years and the majority of subjects in all subgroups (79.7%) were aged 6–12 years. The majority of subjects were male (87.0%) and Caucasian (71.2%). Before study entry, 57.9% of subjects had received prior central nervous system (CNS) medications (de novo, 77.9%; prior placebo, 44.3%; prior aripiprazole 53.4%). The most commonly used CNS medications (>10% of subjects) before study enrollment were antipsychotics (24.5%), psychostimulants (17.9%), anxiolytics (17.6%), and antidepressants (16.7%). Baseline mean (standard deviation [SD]) ABC Irritability subscale scores at the start of the open-label treatment phase were 23.2 (8.9) in de novo subjects (n = 80), 21.5 (9.8) in prior placebo subjects (n = 68) and 15.0 (9.2) in prior aripiprazole subjects (n = 166).
Aripiprazole dosing
Overall, 200 subjects received at least one dose of aripiprazole during the last week of the study; the mean daily dose of aripiprazole during the final week of treatment (week 52) in these subjects was 10.6 mg/day (range: 1.1–15.0 mg/day). The mean daily dose of aripiprazole on the last day of dosing for all subjects, including those who discontinued prematurely, was 9.6 mg/day (range: 1.1–15 mg aripiprazole daily). At week 52, the distribution of aripiprazole dosing was as follows: 0 mg/day, n = 1 (0.5%); 2 mg/day, n = 10 (5.0%); 5 mg/day, n = 35 (17.5%); 10 mg/day, n = 64 (32.0%); 15 mg/day, n = 90 (45.0%) (based on the most frequent dose received during week 52; in case of ties, the highest dose was used).
Concomitant medications
At their last study visit, 38.2% of subjects were receiving concomitant CNS medications; the most commonly used medications included antidepressants (13.4%), psychostimulants (11.5%), and antiepileptics (5.9%). For subjects who completed the study (n = 199), the most commonly used CNS medications (excluding analgesics and antipyretics) were anxiolytics (20.1%), antidepressants (16.6%), psychostimulants (15.1%), antiepileptics (6.5%), and anticholinergics (6.0%).
Fifteen subjects (4.5%) took concomitant medication for the treatment of EPS at some point during the study. These included anticholinergics (3.9%), beta-blocking agents (0.9%), and dopaminergic agents (amantadine; 0.3%). In addition, 114 subjects (34.5%) received concomitant antihistamine medication (including diphenhydramine) during the study; most commonly for the treatment of adverse events (19.4%).
Efficacy assessments
Prior placebo and de novo subjects
Baseline ABC-I and CGI-S scores were slightly higher in de novo subjects than prior placebo subjects. The mean change in ABC Irritability subscale scores by study week for subjects continuing with aripiprazole treatment is shown in Figure 1. For prior placebo subjects and de novo subjects, improvement in ABC-I scores occurred early in the course of treatment and was maintained to study endpoint. At week 52 (observed case [OC] data set), the mean change from baseline ABC Irritability scores was −6.1 (11.9) and −8.0 (10.1) in placebo and de novo subjects, respectively. Similar improvements in ABC Irritability scores were seen for the last observation carried forward (LOCF) data set at week 52 (Fig. 1). In the antecedent placebo-controlled trials, the mean baseline ABC-I scores for the subjects randomized to placebo (the “prior placebo” subjects in this trial) were 26.9 and 30.8 in the fixed-dose and flexible-dose studies, respectively, and mean improvement in ABC-I scores was 8.4 and 5.0 points after 8 weeks of double-blind treatment.

Mean change in ABC Irritability subscale scores by week (OC data set, also showing LOCF endpoint), efficacy sample. ABC Irritability subscale scores at baseline: de novo, 23.2 (8.9); prior placebo, 21.5 (9.8); prior aripiprazole, 15.0 (9.2). Prior aripiprazole subjects had previously received aripiprazole for 8 weeks before open-label baseline assessments; thus, lower baseline values in this group reflect less severe impairment at baseline and mean change scores represent the change during long-term treatment, subsequent to improvement experienced during double-blind treatment. ABC = Aberrant Behavior Checklist; LOCF = last observation carried forward; OC = observed case; n = Week 52 OC patient numbers (week 52 LOCF patient numbers).
The mean changes in CGI-S scores for these subgroups are shown in Table 2. Both prior placebo and de novo subgroups showed mean improvements from baseline in CGI-S scores at week 52 (OC) and endpoint (LOCF).
For the Pediatric Quality of Life Inventory combined total scores, positive scores signify improvement.
Prior aripiprazole subjects had previously received aripiprazole for 8 weeks before open-label baseline assessments; thus, lower baseline values in this group reflect less severe impairment at baseline and mean change scores represent the change during long-term treatment, subsequent to improvement experienced during double-blind treatment.
CY–BOCS = Children's Yale–Brown Obsessive Compulsive Scale; LOCF = last observation carried forward; OC = observed case; SD = standard deviation.
Mean (SD) CGI-I scores showed that symptom improvement was maintained over the 52-week treatment period for prior placebo (2.4 [1.2]) and de novo (2.7 [1.3]) subjects (week 52; LOCF). At endpoint, the majority of prior placebo and de novo subjects had a CGI-I score of much improved or very much improved compared with their condition before treatment.
Mean (SD) change from baseline in additional efficacy outcomes at week 52 (OC) and endpoint (LOCF) is also shown in Table 2. Both prior placebo and de novo subject groups showed a mean improvement on ABC Social Withdrawal, Stereotypic Behavior, Hyperactivity, and Inappropriate Speech subscale scores at week 52 (OC) and endpoint (LOCF).
Prior placebo and de novo subjects showed a mean improvement from baseline in CY-BOCS Compulsion Scale score at week 52 (OC) and endpoint (LOCF) and showed increases in PedsQL scores from baseline to endpoint, indicating improvement in quality of life. Mean change in CGSQ scores at endpoint (LOCF) showed improvement in caregiver strain in the prior placebo and de novo subject groups.
Prior aripiprazole subjects
For prior aripiprazole subjects, ABC Irritability scores remained constant over the course of long-term treatment, indicating that prior improvement experienced with aripiprazole treatment during the randomized treatment period was maintained during the 52-week open-label treatment period; at week 52 (OC data set), the mean change from baseline in ABC Irritability scores was + 0.7 (10.2). In the antecedent placebo-controlled trials, the mean baseline ABC-I scores for the subjects randomized to aripiprazole (the “prior aripiprazole” subjects in this trial) were between 28 and 30 at the original baseline at the start of double-blind treatment, and there was a mean improvement in ABC-I scores of 12 to 14 points after 8 weeks of double-blind treatment.
CGI-S scores at week 52 (OC) and endpoint (LOCF) were generally unchanged (Table 2), although the mean CGI-S score at open-label baseline was 3.9 points, compared with a mean baseline score of around 5 at baseline in the antecedent placebo-controlled trials. The mean (SD) CGI-I score was 2.5 (1.2) for the prior aripiprazole group at week 52 (LOCF), indicating that symptom improvement was maintained. Distribution of CGI-I scores (Fig. 2) showed that the majority of prior aripiprazole subjects had a CGI-I score of much improved or very much improved at endpoint.
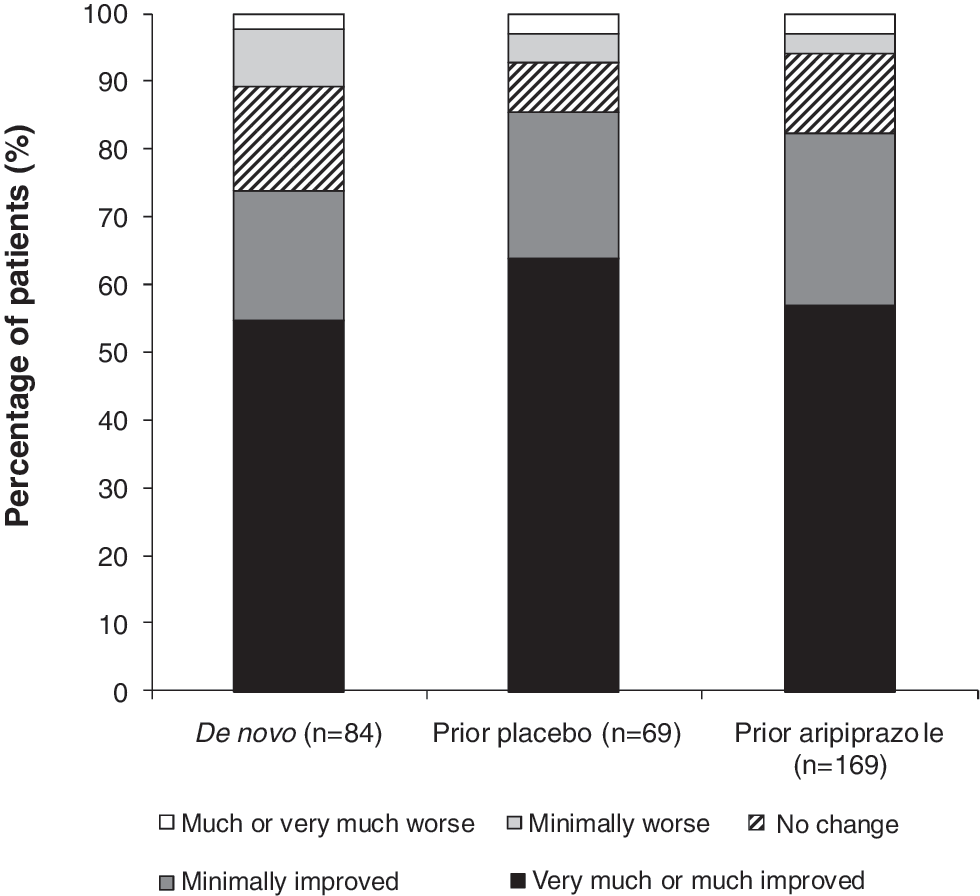
Distribution of CGI-I score at endpoint (LOCF), efficacy sample. CGI Improvement was rated relative to the symptoms at open-label baseline for de novo subjects and relative to double-blind treatment baseline for prior aripiprazole/placebo subjects. CGI = Clinical Global Impressions.
For all ABC subscales, open-label baseline values for the prior aripiprazole group were lower than for the prior placebo and de novo subject groups, as would be expected given that prior aripiprazole subjects had previously received active treatment for 8 weeks (Table 2). Prior aripiprazole subjects showed a mean improvement in ABC Social Withdrawal, Stereotypic Behavior, Hyperactivity, and Inappropriate Speech subscale scores at week 52 for patients who remained on treatment (OC data). Mean changes in ABC Social Withdrawal, Stereotypic Behavior, Hyperactivity, and Inappropriate Speech subscale scores for prior aripiprazole subjects at endpoint (LOCF) were small.
Prior aripiprazole subjects showed a slight mean improvement from baseline in CY-BOCS Compulsion Scale score at week 52 (OC) and endpoint (LOCF). Prior aripiprazole subjects experienced mean increases in PedsQL scores from baseline to endpoint, indicating improvement in quality of life, although no overall improvement in CGSQ scores at endpoint (LOCF) was seen for prior aripiprazole subjects.
Discussion
Results from this long-term open-label study suggest that aripiprazole, for up to 1 year, can reduce symptoms of irritability in children and adolescents with autistic disorder who also demonstrated behaviors such as irritability, agitation, and self-injurious behavior or a combination of these. Symptoms of irritability, assessed using the caregiver-rated ABC Irritability subscale score, were improved early in the course of treatment and these improvements were maintained over the 52-week study period in patients receiving aripiprazole for the first time during this study (prior placebo subjects and the majority of de novo subjects). The pattern of improvement seen in the first few weeks of aripiprazole treatment for placebo rollover and de novo subjects was similar to that observed with aripiprazole in the double-blind studies: early symptom improvement within the first few weeks followed by further improvement up to week 8 (Marcus et al. 2009; Owen et al. 2009). For individuals who had previously received 8 weeks of aripiprazole treatment during the prior double-blind studies, improvements in irritability symptoms were maintained over the 52-week treatment period. However, it should be considered that this study was not specifically designed to assess maintenance of effect or the prevention of relapse. Consistent with improvement in symptoms assessed using the caregiver-rated ABC Irritability subscale scores, the distribution of clinician-rated CGI-I score at endpoint suggests that aripiprazole improved symptoms of irritability for up to 52 weeks in the majority of patients. Further, completion rates in this study were high, with 60% of subjects finishing 52 weeks of treatment and few subjects (6.1%) discontinuing long-term treatment due to lack of efficacy. Discontinuation rates due to lack of efficacy were also low in the double-blind studies; no aripiprazole-treated subjects in the fixed-dose study (Marcus et al. 2009) and 2.1% of aripiprazole-treated subjects in the flexible-dose study discontinued treatment due to lack of efficacy (Owen et al. 2009).
In addition to the improvements in irritability observed in this long-term study, prior placebo and de novo subjects also demonstrated improvements on the other ABC subscales. It is also interesting to note that aripiprazole treatment resulted in improvements in quality of life measures at endpoint in these groups, as measured on the PedsQL and CGSQ scales, suggesting that improvements in irritability associated with autistic disorder during 52 weeks of treatment may translate into improved day-to-day functioning for the children and their families. Improvement in these measures was also seen in subjects receiving aripiprazole during the short-term studies (Marcus et al. 2009; Owen et al. 2009), and this improvement was maintained over the 52-week treatment period in this study.
The safety and tolerability findings from this study are reported elsewhere (Marcus et al. In press). Briefly, aripiprazole was generally well tolerated as a long-term treatment in this population; discontinuations due to adverse events were 10.6%, and the most common AEs leading to discontinuation were aggression and weight increase (Marcus et al. In press). The most frequently reported AEs (occurring in ≥ 10% of subjects) in the total study population during long-term treatment were weight increase (23%), vomiting (19%), nasopharyngitis (13%), increased appetite (13%), pyrexia (12%), upper respiratory tract infection (12%), and insomnia (10%). EPS-related AEs occurred in 15% of subjects (Marcus et al. In press). Nine (2.7%) subjects reported serious AEs; only aggression was reported for more than one subject (Marcus et al., In press). Increased weight gain, though present, appeared to plateau over time. As with all atypical antipsychotics, individuals receiving treatment with aripiprazole should be monitored for weight gain, and this should be proactively managed should it occur (Correll 2008). As with all pharmacologic treatment generally, the decision to initiate medication treatment in pediatric patients with irritability associated with autistic disorder should be made between healthcare providers and caregivers only after a thorough diagnostic evaluation and discussion of both the benefits and the risks.
The findings of this study are strengthened by the large patient population that was enrolled and the relatively high numbers of subjects who continued to receive aripiprazole for the 52-week study duration. However, the efficacy findings should be interpreted with consideration of several limitations such as the open-label study design, along with the challenges of interpreting long-term study data. Further, this study permitted the use of a range of concomitant psychotropic and nonpsychotropic medications. At their last visit, 38.2% of subjects were receiving a concomitant CNS medication, most frequently an antidepressant (13.4%), a psychostimulant (11.5%), or an antiepileptic (5.9%). While this concomitant medication use is more likely to reflect real-life clinical practice, it is important to recognize the rate of concomitant medication use when interpreting the efficacy findings reported. The rate of concomitant medication use for the treatment of EPS reported here (5%) was similar to that reported in subjects receiving aripiprazole in the short-term fixed-dose (6%) (Marcus et al. 2009) and flexible-dose studies (2%) (Owen et al. 2009).
Additional limitations should also be considered. First, all patients were re-titrated to aripiprazole 2 mg/day at entry into open-label treatment. While this may have had an impact on prior aripiprazole patients who were receiving higher aripiprazole doses during the previous studies, we did not systematically evaluate the effects of a reduction in dose to 2 mg/day. It was necessary to dose in this manner, as the treatments patients were receiving during double-blind treatment were not known at entry into open-label treatment. This necessitated re-titration to therapeutic dose.
Nevertheless, the long half-life of aripiprazole (∼75 hours) makes discontinuation symptoms somewhat unlikely during re-titration. It is worth noting, however, that the majority of subjects in this study were taking 10 or 15 mg/day at the end of the study. Second, the majority of subjects in these studies were male, as would be expected based on the higher prevalence of autistic disorder in males than in females. As such, the generalizability of our findings to a predominantly female population is unknown. Third, a washout period of 4 days was used in this study, as some patients could potentially decompensate substantially with longer washout periods. Although the washout period for prior medications used may be considered too short for previous antipsychotic washout, the first ABC-I assessment was at week 4, by which time all prohibited antipsychotics would have been cleared. Fourth, as this study included only subjects with autistic disorder, the generalizability of these results to other pervasive developmental disorders is unknown. Finally, as this was not a comparison study, judgments regarding the relative efficacy of aripiprazole compared with other antipsychotics in this population cannot be made.
Conclusions
Aripiprazole reduced symptoms of irritability associated with autistic disorder in pediatric subjects aged 6–17 years who were studied for up to 1 year.
Clinical Significance
For chronic conditions that may require long-term treatment, there is a need for data regarding treatment effects that go beyond the acute phase of an illness to help inform clinical decision making. The results from this study confirm that there can be continued benefit—for up to 1 year—from the use of aripiprazole for the treatment of the symptoms of irritability associated with autistic disorder in pediatric subjects aged 6 to 17 years.
Footnotes
Disclosures
Michael Aman was a consultant to and received grant support from Bristol-Myers Squibb.
Lisa Kamen, Raymond Mankoski, George Manos, and Ronald N. Marcus are employees of Bristol-Myers Squibb.
Patricia Corey-Lisle and Randall Owen are former employees of Bristol-Myers Squibb.
Robert D. McQuade and William H. Carson are employees of Otsuka Pharmaceutical Development & Commercialization, Inc.
Acknowledgments
This study was supported by Bristol-Myers Squibb (Princeton, NJ) and Otsuka Pharmaceutical Co., Ltd. (Tokyo, Japan). Editorial support for the preparation of this article was provided by Ogilvy Healthworld Medical Education; funding was provided by Bristol-Myers Squibb.
Clinical trials information: NCT00365859; registry: