
Abstract
Objective:
The aim of this study was to evaluate the tolerability and effectiveness of the methylphenidate transdermal system (MTS) over 6 months in adolescents with attention-deficit/hyperactivity disorder (ADHD).
Methods:
This was an industry-sponsored, 30-center, open-label study of subjects aged 13–17 years with ADHD. Subjects were dose-optimized with MTS (10–30 mg/9 hours) over 5 weeks and then dose-maintained for up to 5 months. Tolerability evaluations included treatment-emergent adverse events (TEAEs) and dermal responses. Effectiveness was assessed with the ADHD-Rating Scale-IV (ADHD-RS-IV).
Results:
A total of 162 subjects received MTS treatment. The majority of TEAEs (>99%) were mild or moderate in intensity, and the most frequently reported TEAE was decreased appetite (15.4%). Thirteen subjects discontinued the study due to TEAEs. The majority (93.6%) of dermatologic reactions indicated mild erythema. There was significant improvement in mean ADHD-RS-IV total scores from study entry to end point (p < 0.001).
Conclusion:
Slightly more than half (54.0%) of subjects completed this 6-month, open-label extension study of MTS; the primary reason for discontinuation was withdrawn consent (36.0%). Reported TEAEs and skin tolerability findings were similar to those observed with MTS use in children and adolescents. MTS treatment resulted in a decrease in ADHD symptoms as rated by clinicians.
NIH Clinical Trials Registry:
# NCT00501293
Introduction
The treatment of ADHD symptoms for adolescents may involve pharmacotherapy along with psychological, educational, and social interventions (Pliszka 2007). Stimulant medications have gained recognition as the most widely used pharmacologic treatment options for ADHD and are recommended as first-line therapy in several practice guidelines (Global ADHD Working Group 2005; Pliszka et al. 2006; Pliszka 2007). Methylphenidate (MPH), a central nervous system stimulant, has been established as an effective agent in the treatment of ADHD and is widely prescribed (Brinker et al. 2007; Castle et al. 2007). In 2006, the U.S. Food and Drug Administration (FDA) approved the MPH transdermal system (MTS), with a wear time of up to 9 hours, for the treatment of ADHD in children aged 6–12 years (Daytrana package insert 2008; Shire Pharmaceuticals website) as an alternative to oral extended-release and oral immediate-release MPH formulation. MTS is a diffusion-based patch that releases racemic MPH continuously on application to the skin (Pelham et al. 2005a; Heal and Pierce 2006). Because the dose of drug delivered depends on the surface area of the patch and the length of time that the patch is worn (Pelham et al. 2005a), MTS offers individuals the ability to tailor the duration of effect and manage adverse events (AEs) by removing the patch. In addition, the patch also offers an option for individuals with ADHD who have difficulty tolerating or swallowing oral medications (Wilens et al. 2008).
Despite a large body of literature on the effectiveness and tolerability of MTS in short- and long-term studies in children (Pelham et al. 2005b; McGough et al. 2006; Findling et al. 2008; Wilens et al. 2008; Findling et al. 2009), there are no published reports on the extended use of MTS beyond 7 weeks in adolescents aged 13–17 years (Findling et al. 2010). Because MTS may offer novel benefits for adolescents not available with oral formulations of MPH, this study, one of the largest adolescent, long-term trials of stimulants, evaluated the tolerability and effectiveness of MTS over 6 months for the treatment of ADHD in adolescents.
Methods
Data were collected at 30 sites across the United States. The institutional review board at each site approved the study. Each subject completed an assent form to participate in the study, and the subject's parent or caregiver provided written consent in accordance with the International Conference on Harmonization Good Clinical Practice Guideline E6 and applicable regulations (European Medicines Agency 2002). The study was conducted over a period of approximately 14 months; the first subject consent was obtained on August 29, 2007, and the last follow-up visit was completed on October 21, 2008.
Subjects
Adolescents aged 13–17 years with a primary diagnosis of ADHD according to the Diagnostic and Statistical Manual of Mental Disorders, 4th edition, Text Revision criteria (American Psychiatric Association, 2000), who had previously participated in a short-term, randomized, double-blind, placebo-controlled, dose-optimization study of MTS (Findling et al. 2010), were eligible to participate in the present trial. In addition, subjects must have completed all required study visits or completed a 5-week dose-optimization period without achieving an acceptable condition (i.e., ≥25% decrease from baseline in a subject's ADHD-Rating Scale-IV [ADHD-RS-IV] score with minimal side effects). Inclusion criteria for the antecedent study incorporated the following: A total score of ≥26 on the ADHD-RS-IV at baseline; an intelligence quotient (IQ) score of 80 or above; electrocardiogram (ECG) results within normal range or variants that were not clinically significant; blood pressure measurements within the 95th percentile for age, gender, and height; and no current skin disease or history of skin disease or other skin problems including sensitive skin or signs of skin irritation. Criteria for exclusion in the antecedent study included: Conduct disorder or co-morbid psychiatric illnesses that contraindicated or could confound MTS treatment responses, or a history of serious cardiac problems, suicidal ideation, substance abuse, or seizures during the previous 2 years. In addition, subjects who were nonresponsive to stimulant medications and those receiving clonidine, atomoxetine, antidepressants, sedatives, antipsychotics, anxiolytics, P450 enzyme-altering agents, or other investigational medications within 30 days prior to antecedent study screening were not eligible to participate.
Subjects were not eligible to participate in the extension study if they were discontinued from the antecedent study due to a protocol violation (including noncompliance), an AE for which continued treatment would be medically contraindicated, or a serious adverse event (SAE). Subjects with considerable general medical illness (except mild, stable asthma) or an unstable medical condition, disability, or other condition the investigator believed might interfere with or prevent completion of the study were also excluded. The use of concomitant therapies that could potentially interfere with the tolerability or effectiveness assessments of MTS were prohibited throughout the trial.
Study design and treatment
This was a multicenter, open-label, extension study evaluating the safety and efficacy of MTS (10-, 15-, 20- or 30-mg/9-hour patches) administered for approximately 6 months to 13- to 17-year-old adolescents with ADHD who received either MTS or placebo transdermal system (PTS) in the antecedent trial. This study consisted of three experimental periods: Dose optimization (5 weekly visits), dose maintenance (5 monthly visits), and a 7-day posttreatment follow up (Fig. 1).
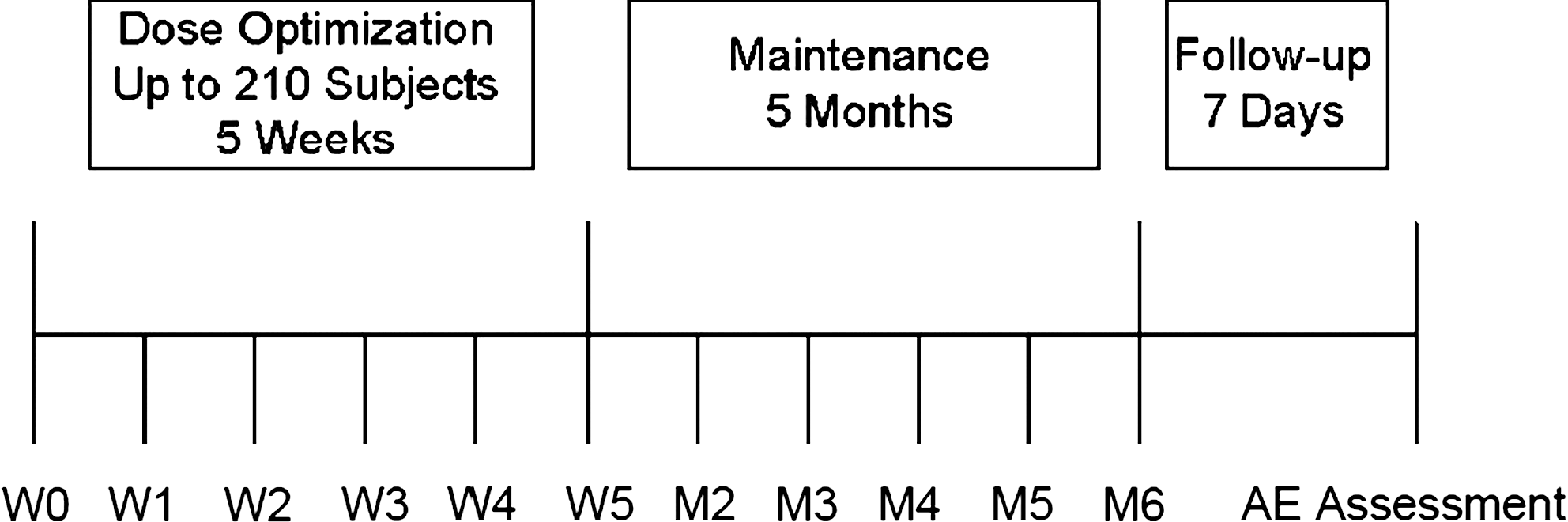
Study design schematic. W = Week; M = month; AE = adverse event.
Regardless of prior treatment in the antecedent study, all subjects began the extension study with 10-mg/9-hour MTS. During the dose-optimization period, beginning at week 1 and extending through week 5, subjects were titrated to an optimal dose of medication. Titration was based on the overall response of the subject defined as “acceptable,” “ineffective,” or “intolerable.” An “acceptable” response to medication was defined as ≥25% decrease from baseline in a subject's ADHD-RS-IV score with minimal side effects. A subject would remain on his or her acceptable-response dose for the remainder of the study unless the clinician believed that the subject could receive further symptom reduction from titration to a higher dose. Subjects who had not reached an acceptable response by end of week 5 were withdrawn from the study. An “ineffective” response was defined as <25% change in a subject's ADHD-RS-IV score from baseline. If the subject had an ineffective response and was tolerating his or her current dose of medication, the subject was to receive the next higher dose strength for the following week. If in the clinician's opinion a more effective response was not achieved at a higher dose, the subject was tapered downward to the previous dose strength. An “intolerable” response was defined as an unacceptable safety profile experienced by the subject. If the subject had an intolerable response, the subject was titrated downward to the previous dose strength. If the lowered dose strength also produced an intolerable effect, the subject was discontinued from the study.
During the dose-maintenance period, beginning at week 6 and extending through month 6, titration (upward or downward) was permissible based on the investigator's assessment of the subject's response and occurrence of AEs, although it was anticipated that subjects would be maintained on their optimized MTS dose between the 5 monthly visits.
Additional tolerability information was collected during a 7-day follow-up period, although no scheduled study visit took place.
MTS patches were applied to the subject's hip each day on awakening (∼7 a.m.) and removed after 9 hours (∼4 p.m.). Patch applications were to be alternated between the left and right hip so that the same site was not used for two consecutive applications. As compliance was assessed by patch counts, subjects were instructed to bring unused MTS patches and partially empty or empty trays to each study visit. A minimum of 80% compliance was required between two consecutive visits during both dose-optimization and dose-maintenance periods for ongoing study participation.
Tolerability assessments
AEs were monitored at each study visit and assessed by an open-ended inquiry along with specific dermatological questions asked by an investigator or qualified evaluator. AEs were considered treatment emergent if they began or worsened on or after application of the first patch and occurred before or at the same time as application of the last patch. All AEs were coded and defined using the Medical Dictionary for Regulatory Activities (MedDRA), version 7.0. The severity and relationship of AEs to study treatment were assessed by the investigators. Signs of skin irritation or symptoms of discomfort were not recorded as AEs unless they occurred at a site different from the patch application site or required pharmacologic treatment.
A physical examination was performed at study entry and at the month 6 visit. Height was measured at study entry and at the month 6 visit. Body weight was recorded at all visits. Vital sign measurements (oral temperature, systolic blood pressure [SBP], diastolic blood pressure [DBP], pulse, and sitting respiratory rate) were obtained at all study visits. A standard 12-lead ECG was performed at entry, week 4, month 3, and month 6. Blood and urine samples were collected for routine clinical laboratory evaluations (hematology, serum chemistry, and urinalysis tests) at entry, week 4, month 3, and month 6. Physical examinations, height, weight, and vital-sign measurements were performed by the investigator. The clinical significance of any vital sign measurement outside of the normal range was determined by the investigator.
Reference ranges for clinical laboratory evaluations were provided by Covance Central Laboratory Services, Indianapolis, Indiana, and were used to assess clinical significance. All ECG data were analyzed by eResearch Technology, Philadelphia, Pennsylvania, and evaluated as normal or abnormal potentially clinically important. The investigator determined, based on normal ranges, whether the ECG was normal, abnormal but not clinically significant, or abnormal and clinically significant. Any changes noted between evaluation data at study entry and data obtained at scheduled visits deemed to be clinically significant by the investigator were considered an AE.
A dermatological evaluation was completed at each study visit to assess skin reactions or irritations, skin discomfort, and transdermal system adherence at the current and prior application site. The dermal response scale (DRS) was used to evaluate observed skin findings: 0 = No evidence of irritation; 1 = minimal erythema; 2 = definite erythema; 3 = erythema and papules; 4 = definite edema; 5 = erythema, edema, and papules; 6 = vesicular eruption; and 7 = strong reaction beyond the site. The experience of discomfort and pruritus scale (EODP) was used to evaluate the overall level of discomfort at the patch application site: 0 = No discomfort; 1 = mild discomfort; 2 = moderate but tolerable discomfort; and 3 = severe, intolerable discomfort. If discomfort was present (score ≥1), then the type of discomfort (burning, itching, or other) was specified. The transdermal system adherence scale (TSA) was used to examine the adherence of the patch at the current application site: 0 = ≥90% Adhered (essentially no lift off the skin); 1 = ≥75% to <90% adhered (some edges only lifting off of the skin); 2 = <50% to <75% adhered (less than half of the system lifting off of the skin); 3 = <50% adhered but not detached (more than half of the system lifting off of the skin without falling off); or 4 = MTS detached (system completely off the skin).
Subjects completed the nonvalidated, 5-item Post-Sleep Questionnaire to record the effects of MTS treatment on time to sleep, sleep latency, frequency and duration of interrupted sleep, duration of sleep, and overall sleep quality. The questionnaire provided the following questions: [item 1] “What time did you go to bed last night (p.m.)?”, [item 2] How long did it take you to fall asleep (min)?”, [item 3] “Did you wake up during the night (yes or no)?”, [item 3a] “How many times did you wake up during the night?”, [item 3b] “How long were you awake during the night (min)?”, [item 4] “What time did you wake up in the morning (a.m.)?”, and [item 5] “Rate the overall quality of sleep last evening (very poor, poor, average, good, or very good)”. The Post-Sleep Questionnaire was administered at entry and at the month 6 visit.
Effectiveness assessments
The primary measure of effectiveness was the ADHD-RS-IV (DuPaul et al. 1998). Other effectiveness measures included the Conners' Parent Rating Scales-Revised Short Form (CPRS-R) (Conners 1997), Clinical Global Impressions-Improvement (CGI-I) (U.S. Department of Health, Education, and Welfare–National Institutes of Mental Health 1976), and Parent Global Assessment (PGA). The ADHD-RS-IV and CGI-I were completed by investigators, and the CPRS-R and PGA were completed by parents or caregivers at each study visit.
The quality of life (QoL) of adolescents participating in the study was assessed using the Youth Quality of Life–Research (Y-QoL-R) instrument (Topolski et al. 2002). Such measures reflect functioning and impairments, both important aspects of ADHD treatment. The Y-QoL-R is a validated 56-item questionnaire consisting of two modules, perceptual (those known only to the individual) and contextual (those that are potentially verifiable). The perceptual module consists of 41 items, categorized into four individual domains (self, relationship, environment, and general QoL). A total of five scores can be generated from the perceptual items, a total score and four individual domain scores. The contextual module consists of 15 items with each item scored individually. The Y-QoL-R scores are transformed to a 0 to 100 scale for easy interpretability with higher scores indicating better QoL. The Y-QoL-R is a self-reported instrument and was completed by the subjects at entry and at month 6.
Statistical analysis
In general, tolerability assessments were analyzed using descriptive statistics and included all subjects who received at least one dose of MTS.
Effectiveness analyses were performed on the intent-to-treat (ITT) population, which included subjects who enrolled, received at least one dose of MTS, and had a least one postentry assessment using the primary effectiveness measure ADHD-RS-IV. Analysis of the mean change in ADHD-RS-IV score to end point from baseline of the antecedent study was performed using summary statistics and 95% confidence intervals. A p value was presented using a paired t-test to assess if the change from baseline of the antecedent study to visit/end point within each treatment of antecedent study was significantly different from zero. End point is the last nonmissing assessment obtained postbaseline. Analysis of the mean change in CPRS-R, CGI-I, PGA, and Y-QoL-R scores to end point from baseline of the antecedent study were performed using summary statistics. Prior to analysis, CGI-I and PGA scores were dichotomized into an improvement category (consisting of “very much improved” and “much improved”) and no improvement category (consisting of “minimally improved”, “no change”, “minimally worse”, “much worse”, and “very much worse”). A paired t-test was calculated for CPRS-R and Y-QoL-R scores as described above for ADHD-RS-IV by antecedent treatment.
Results
Study population
Of the 217 subjects enrolled in the antecedent study, 57.1% (124/217) of subjects completed the 7-week trial and of these, 93.5% (116/124) enrolled into the current study. Of note, 42.9% (93/217) of subjects did not complete the antecedent trial. Lack of efficacy (51.6%, 48/93) and protocol violations (20.4%, 19/93) were the greatest contributing factors to antecedent study discontinuation. Of subjects that did not complete the antecedent trial, 50.5% (47/93) were eligible (per inclusion and exclusion criteria) to enroll into the current study. Therefore, a total of 163 subjects were enrolled into this study (Fig. 2). Among these subjects, 110 previously received MTS and 53 received PTS in the antecedent trial. Overall, 88 (54.0%) completed the current study. Of the 75 subjects who did not complete the study, the most common reasons for early discontinuation were withdrawn consent (27/75, 36.0%) and lost to follow up (19/75, 25.3%). Subjects also discontinued as the result of AEs (13/75, 17.3%), lack of efficacy (5/75, 6.7%), protocol violations (3/75, 4.0%), and other reasons (8/75, 10.7%). Of enrolled subjects, 162 received at least one dose of MTS and were included in the safety population; 1 subject failed to receive MTS postentry. A total of 4 subjects in the safety population were excluded from the intent-to-treat (ITT) population because they did not have at least one postentry ADHD-RS-IV assessment.
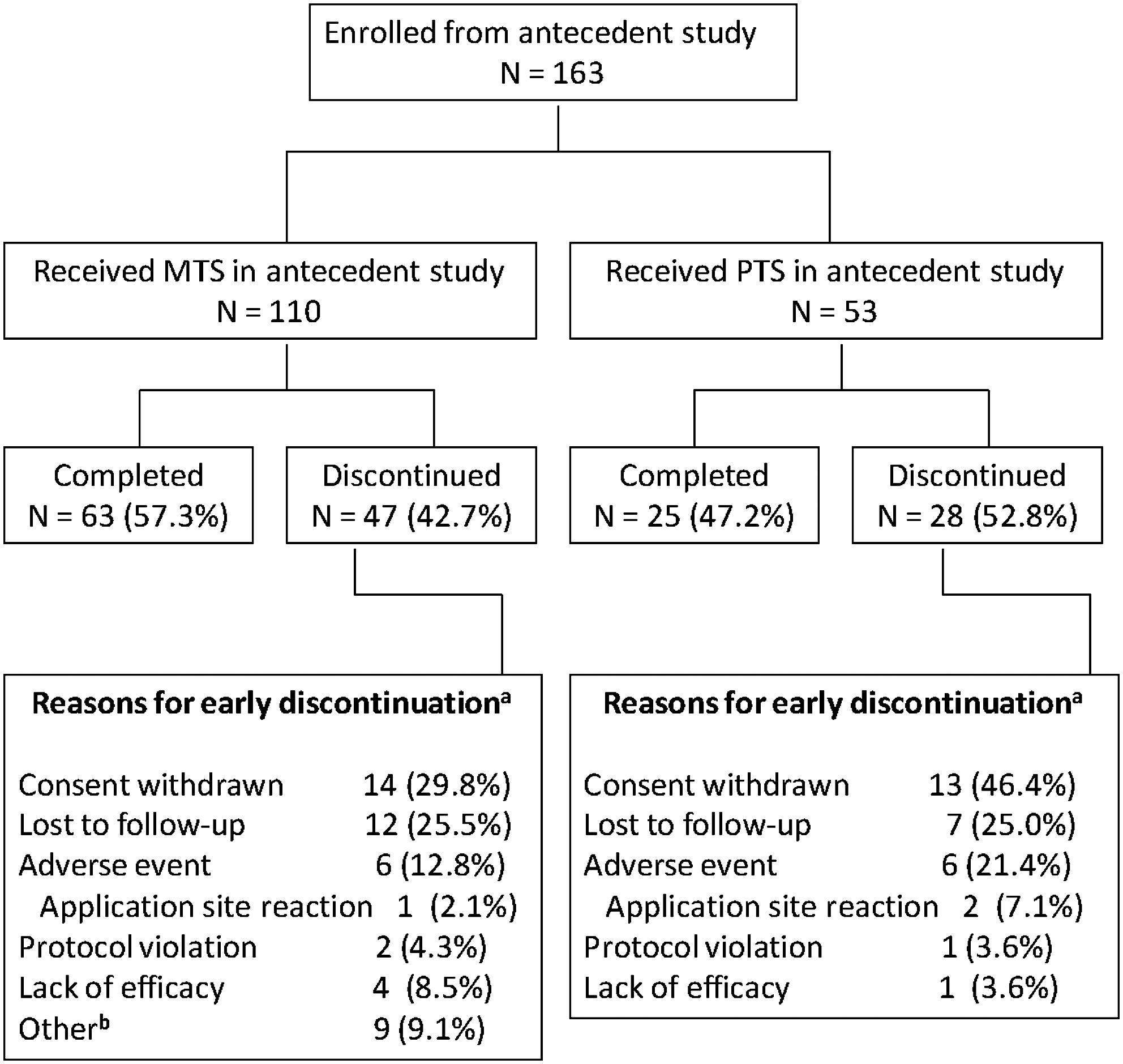
Disposition of subjects through the extension study. aPercentages are based on the number of enrolled subjects who did not complete the 5-month dose-maintenance period of the study. b“Other” reasons include 5 subjects who were noncompliant with study medication, 1 subject who was enrolled in error, 1 subject with an application site reaction that was not recorded as an adverse event, 1 subject who went into a rehabilitation program, and 1 subject who felt the patch was a nuisance and refused to continue. Note: Subjects may have more than one reason for termination.
The demographic and baseline characteristics for the safety population are displayed in Table 1. The majority of subjects were male (121/162, 74.7%) and were white (126/162, 77.8%), with a mean (SD) age of 14.5 (1.24) years. A majority (>95%) of subjects in the antecedent study had been titrated to MTS doses of 15, 20, and 30 mg and were required to reinitiate titration from 10-mg MTS with no dose tapering. At the end of the dose-optimization period, most subjects had progressed to the MTS 15- (14/141, 9.9%), 20- (34/141, 24.1%), or 30-mg (85/141, 60.3%) dose.
Demographic and baseline characteristics are summarized based on the screening visit from the antecedent study.
Baseline values are used for weight.
Abbreviations: MTS = methylphenidate transdermal system; PTS = placebo transdermal system; SD = standard deviation.
In this study, of the 5 subjects who discontinued due to lack of efficacy, 2 subjects discontinued at week 5, the cutoff date for achieving an acceptable response to MTS. At month 6, 5.6%, 7.9%, 32.6%, and 53.9% of subjects were receiving MTS doses of 10, 15, 20, and 30 mg, respectively. The median duration of exposure to MTS during the extension study was 168.0 days (range, 3–200 days).
Adverse events
Overall, 336 TEAEs were reported in 119 of 162 subjects (73.5%) in the safety population during the study period; 196 events were reported by 100 subjects during the dose-optimization period and 140 events were reported by 72 subjects during the dose-maintenance period. Of the 336 TEAEs reported, 128 were considered treatment-related, and these TEAEs occurred in 68 (42.0%) subjects.
A majority (334/336, 99.4%) of these TEAEs were mild to moderate in intensity; two events (viral gastroenteritis and application site burning) were considered severe. The event of viral gastroenteritis was considered not related to study medication and resolved during MTS treatment. The event of application site burning was considered related to MTS treatment and led to the subject's discontinuation from the study; the event did not require pharmacologic treatment and resolved in the subject 2 days after study participation ended.
The most frequently reported TEAEs included decreased appetite (15.4%) and headache (11.7%). Of subjects experiencing decreased appetite, 1 subject required a decrease in dose, 1 subject required study medication to be withheld, and 1 subject discontinued from the study. Of subjects experiencing headache, 1 subject required study medication to be withheld, and no subject discontinued from the study. Other commonly reported TEAEs included upper respiratory tract infection (10.5%), nasopharyngitis (8.0%), and irritability (6.2%). Among these events, the frequency of decreased appetite, headache, irritability, and upper respiratory tract infection TEAEs was greater for the antecedent PTS group than the antecedent MTS group. TEAEs are summarized by antecedent treatment group in Table 2.
Abbreviations: MTS = methylphenidate transdermal system; PTS = placebo transdermal system.
No subjects died during the 6 months of study. A total of five SAEs were reported by 4 subjects; one event each of visual hallucination, auditory hallucination, clonic convulsion, syncope, and grand mal convulsion. Of these SAEs, the visual and auditory hallucinations occurring in 1 subject, who previously received MTS in the antecedent study, were the only SAEs considered to be related to treatment. The subject reported having heard voices and experienced a single episode of visual hallucinations for a period of 3 days after which time study medication was withdrawn. The subject was admitted to a hospital and did not experience any further hallucinations before discharge. The event was considered resolved but ultimately led to study discontinuation.
Clonic convulsion occurred in 1 subject who previously received MTS in the antecedent study. The subject was hospitalized, and a computed tomography scan revealed an undiagnosed cavernous hemangioma, suggesting a previous hemorrhage that predated study participation. Study medication was withdrawn, treatment with oxcarbazepine was initiated, and the subject was discharged from the hospital after 3 days with no further incidence of seizures. The subject's mother reported that the subject had been experiencing daily seizures with vomiting over the previous year that had been incorrectly identified as symptoms of a food allergy. The investigator considered the event not related to study treatment but rather the progression of a preexisting condition, and the subject was discontinued.
Syncope occurred in 1 subject who previously received PTS in the antecedent study. The event was preceded by dizziness and occurred during a migraine headache; it was later determined the subject had not eaten breakfast that day. Study treatment was withheld for 1 day and resumed with no further incidence of syncope. All ECGs for this subject were reviewed by a pediatric cardiologist with no findings indicative of structural heart disease or accessory conduction pathway.
The event of grand mal convulsion in 1 subject, who previously received MTS in the antecedent study, occurred after the subject completed study participation. Following completion of the study, the subject was prescribed 10 mg oral MPH and 6 days later the subject experienced a seizure while playing video games. The subject was admitted to the hospital and underwent electroencephalographic examination. Results indicated mild abnormalities, but revealed no epileptic activity. Results from all other examinations (magnetic resonance imaging and computed tomography scans, ECGs, and evaluations of neurological data) were unremarkable.
A total of 13 (8.0%) subjects experienced 19 AEs that ultimately led to study discontinuation. Of these 19 AEs, 17 were considered treatment emergent. One subject each (0.6%) discontinued due to nausea, clonic convulsion, anxiety, compulsions, auditory and visual hallucination, and social avoidant behavior; 2 subjects each (1.2%) discontinued due to affect lability and irritability; and 4 subjects (2.6%) discontinued as a result of an application site–related TEAE (one report each of burning; rash; erythema, edema, and pruritus; and reaction at the application site).
Clinical laboratory evaluations, physical examinations, and vital sign assessments
As judged by the investigator, there were no clinically significant findings between laboratory evaluation parameters (i.e., hematology, serum chemistry, or urinalysis) obtained postentry relative to screening values obtained at the antecedent study.
In general, subjects gained both height and weight during the study. Mean (SD) weight increased by 1.72 (7.984) pounds and height increased 0.82 (0.981) inches from baseline of the antecedent study. Overall, there was a very small change in mean (SD) body mass index (BMI) from baseline of the antecedent study through end point (0.05 [1.428]). Height, weight, and BMI values were converted to z-scores for age based on the Centers for Disease Control and Prevention growth charts (Centers for Disease Control and Prevention 2009). Compared with baseline of the antecedent study, the mean (SD) z-scores at end point were similar for both weight (0.41 [0.933] versus 0.53 [0.919]) and BMI (0.32 [0.965] versus 0.34 [0.992]) and increased for height (0.19 [0.967] versus 0.46 [1.037]).
At all study visits, mean pulse rate was increased from baseline of the antecedent study with a mean (SD) increase of 5.2 (12.84) beats per minute (bpm) and a final value of 79.1 (11.93; range 52–111) bpm at end point. One subject receiving a 30-mg MTS reported an AE of intermittent tachycardia during dose optimization, resulting in a maximum elevated pulse of 128 bpm at the week-4 measurement. The subject's pulse rate decreased to 102 bpm at week 5 and was considered to be within the normal range (<100 bpm) at 86 bpm at month 3. The event was considered to be of mild intensity, related to treatment, and it required no change in dose strength. Changes in SBP and DBP from baseline of the antecedent study were observed at all visits throughout this study. At end point, the mean (SD) increase from baseline of the antecedent study was 4.2 (10.59) mmHg and 1.7 (8.59) mmHg for SBP and DBP with final values of 115.8 (11.25; range 89–156) mmHg and 68.3 (7.72; range 49–89) mmHg, respectively. One subject receiving 30 mg MTS had two reports of AEs of increased blood pressure (BP) related to SBP observations during dose maintenance. At the time of the first event, the subject experienced an elevated BP of 150/76 mmHg at week 5. The subject's BP returned to the normal range at month 4. The event was considered to be of mild intensity, related to treatment, and required no change in dose strength. In the second occurrence, considered mild in intensity and related to treatment, the subject experienced an elevated BP of 156/77 mmHg at month 6 that was unresolved and ongoing at study end point.
Electrocardiogram assessments
During the study, 18.5% (30/162) of subjects had potentially clinically important (PCI) abnormal ECG parameters as reported by the central laboratory; however, none of the abnormalities was considered clinically significant by investigators. At end point, the mean (SD) increase in heart rate from antecedent baseline was 5.9 (13.40) bpm with a final value of 76.6 (12.88; range 51–114) bpm. A total of 7 subjects had PCI values for heart rate (≤50 bpm or ≥100 bpm). No clinically meaningful changes in PR and QRS intervals were observed; mean (SD) values at end point were 140.5 (17.83; range 101–197) msec and 88.3 (9.32; range 68–122) msec, respectively. At end point, no subject had PR intervals that were considered PCI; however, 2 subjects had QRS intervals that were considered PCI (≥120 msec). Decreases in QT interval duration from antecedent study baseline were observed at all assessments during the study, and at end point the mean (SD) decrease in QT interval duration was −7.4 (26.26) msec with a final value of 366.0 (26.77; range 306–439) msec. No subject had QT intervals that were considered PCI. At end point the mean (SD) increase in QTcF interval duration, corrected using the Fridericia formula, from antecedent study baseline was 2.0 (16.59) msec with a final value of 394.7 (19.78; range 342–455) msec. Two subjects had QTcF intervals that were considered PCI (≥450 msec). No subject discontinued the study as the result of a cardiovascular-related AE.
One subject had a conduction disorder (accelerated AV node conduction) with onset prior to study entry. At baseline of the antecedent study, ECG PR intervals for this subject ranged from 91 to 105 msec with no associated QRS abnormalities. The event was considered ongoing at end point of the antecedent study. Although the conduction disorder was classified as an AE, it did not lead to early termination from the antecedent study. Thus, the subject met the inclusion criteria for the current study and was subsequently enrolled without the patient's enrollment being considered a protocol violation. At the end of this study (month 6), the subject's ECG PR interval was 119 msec and the event was considered to be resolved. No potentially clinically important ECG results were recorded during the subject's participation in this study and all ECGs were considered to show no clinically significant abnormalities.
Dermatological evaluations
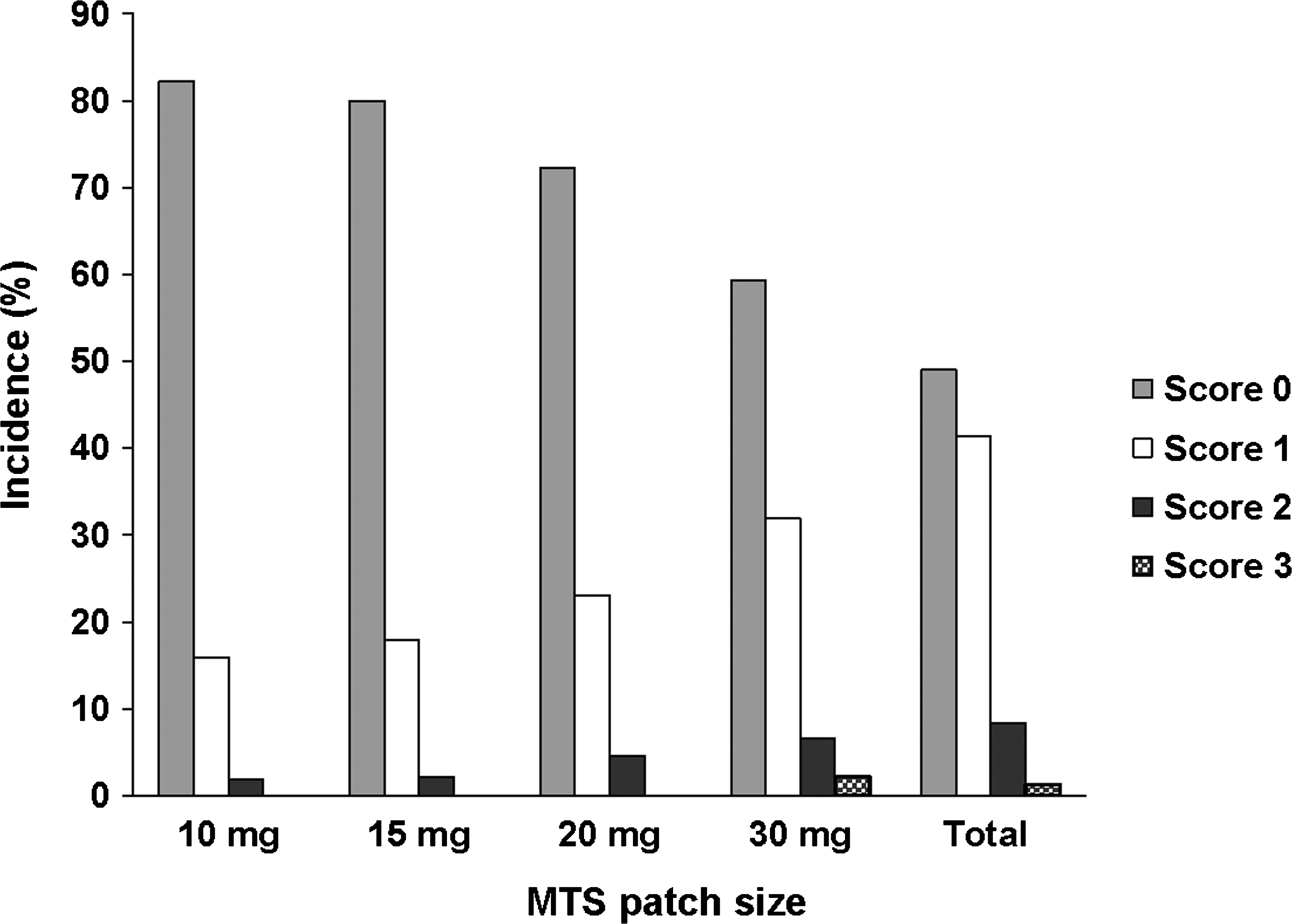
In an analysis of maximum DRS scores, the majority of subjects (83.5%) had a maximum DRS score of 1, 2, or 3, indicating the presence of, at most, erythema with papules at current patch application sites (Fig. 3). No subject had a maximum DRS score >5 at either the current or prior patch application site (Fig. 4). In a similar analysis of maximum EODP scores, the majority of subjects (98.7%) had maximum EODP scores ≤2 at the current patch site during the study (Fig. 5). Two subjects experienced an EODP score of 3 (severe, intolerable discomfort) at the current patch site while receiving 30 mg MTS; of these, 1 subject discontinued the study due to a severe event of application site burning. Four subjects discontinued as a result of an application site reaction; 1 subject was receiving 20 mg MTS and the other 3 subjects were receiving 30 mg MTS.
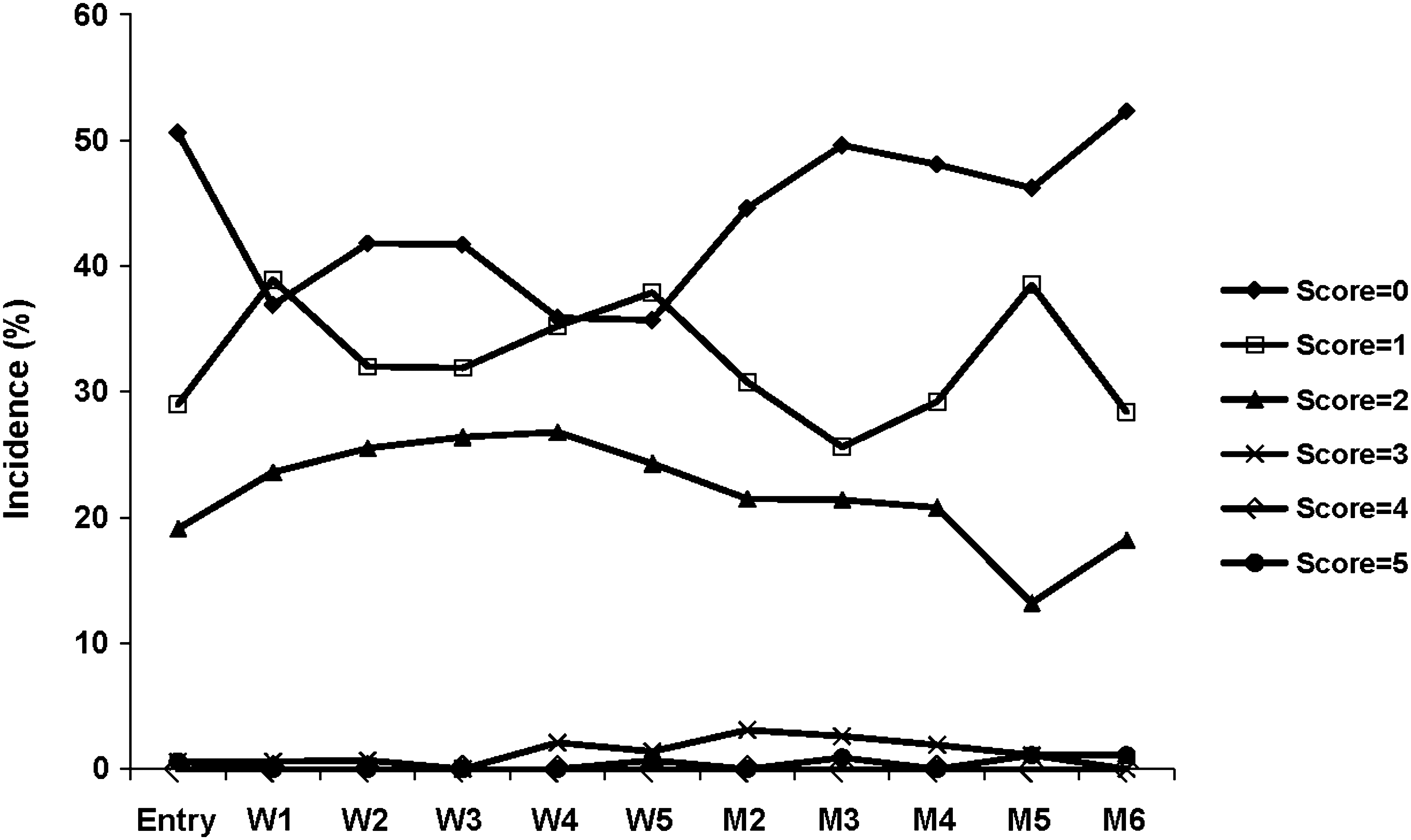
Mean dermal response at the current application site, safety population. Dermal response scale scores range from 0 to 7, where 0 = no evidence of irritation, 1 = minimal erythema, 2 = definite erythema, 3 = erythema and papules, 4 = definite edema, 5 = erythema, edema, and papules, 6 = vesicular eruption, 7 = strong reaction beyond site. Scores of 6 and 7 are not included in the key because no subject had a score greater than 5. W = Week; M = month.
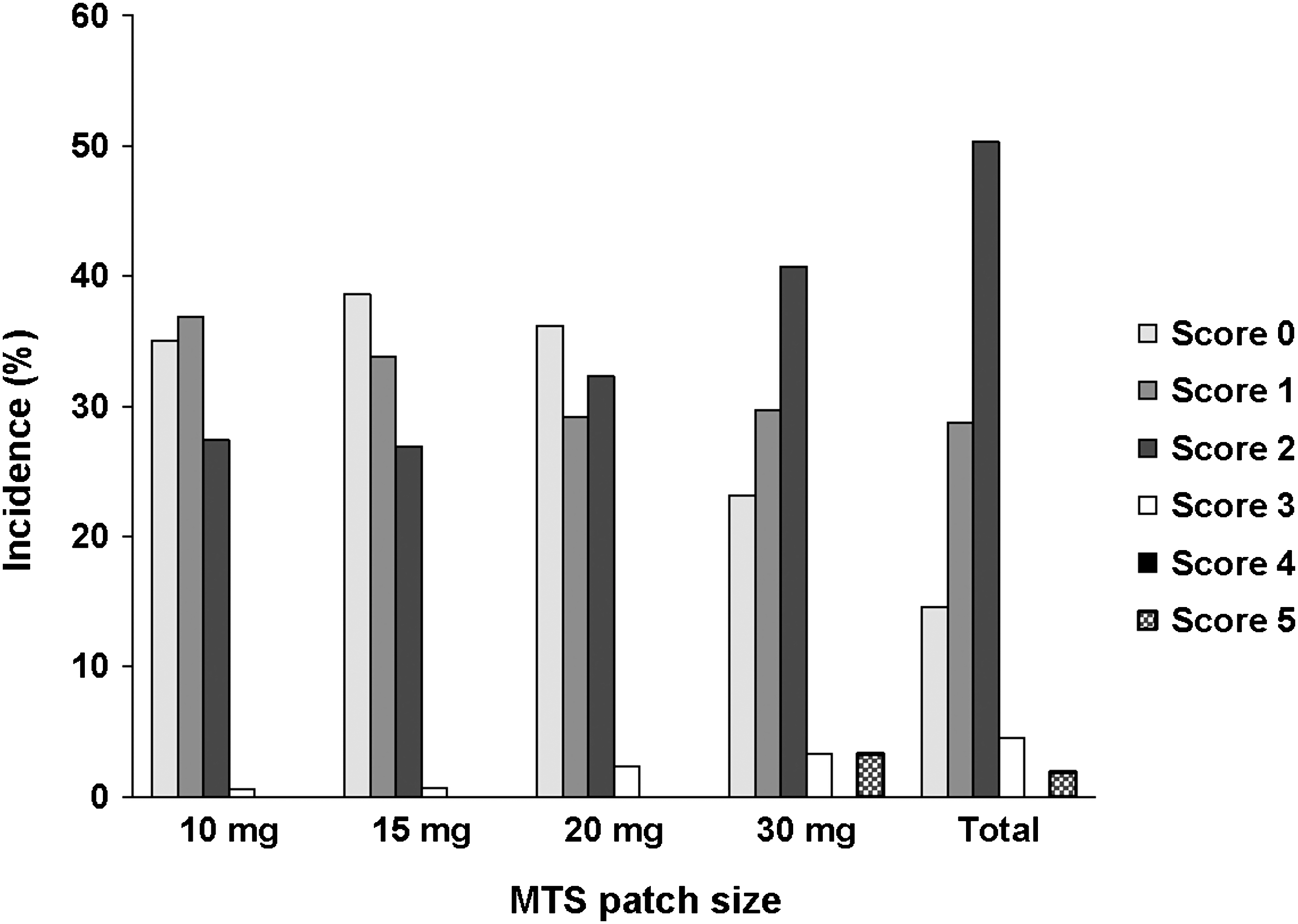
Distribution of maximum dermal response scale (DRS) scores for each patch size, safety population. The DRS was used to evaluate observed skin findings: 0 = no evidence of irritation, 1 = minimal erythema, 2 = definite erythema, 3 = erythema and papules, 4 = definite edema, 5 = erythema, edema, and papules, 6 = vesicular eruption, 7 = strong reaction beyond site. Scores of 6 and 7 are not included because no subject has a score greater than 5. MTS = methylphenidate transdermal system.
Experience of Discomfort and Pruritus (EDOP) scale maximum patch scores at the current application site, safety population. The EDOP scale was used to evaluate the overall level of discomfort at the patch application site: 0 = no discomfort, 1 = mild discomfort, 2 = moderate but tolerable discomfort, 3 = severe, intolerable discomfort. MTS = methylphenidate transdermal system.
The majority of subjects (90.3%) reported a TSA score of 0 (≥90% adhered, essentially no lift off the skin) or 1 (≥75% to <90%, some edges only lifting off of the skin) over the course of the study. Only 5 subjects had a TSA score of 4 (MTS detached, system completely off the skin) at any timepoint during the study.
Post-Sleep Questionnaire
Self-reports on items in the Post-Sleep Questionnaire were similar at all assessment periods. The majority of subjects reported going to bed between 10:00 p.m. and 12:00 a.m. at baseline of the antecedent study (64.1%) and month 6 (61.3%). There were no major differences with regard to sleep latency, and the mean (SD) time in minutes to fall asleep were 23.6 (24.11) and 21.1 (31.69) at baseline of the antecedent study and month 6, respectively. The number of subjects reporting no waking during the night increased throughout the study from 51.5% at baseline of the antecedent study to 66.7% at month 6. The mean (SD) number of awakenings during the night was similar for all subjects, 1.3 (1.36) at baseline of the antecedent study and 1.0 (1.84) at month 6. For subjects who awoke during the night, the mean (SD) duration in minutes of waking was greater at baseline of the antecedent study (13.2 [27.34]) than at month 6 (7.5 [16.18]). The mean (SD) duration of sleep in hours was similar at baseline of the antecedent study (8.22 [1.543]) and month 6 (8.59 [1.684]). There were no major differences in the percentages of subjects rating overall sleep quality as either “good” or “very good” at baseline of the antecedent study (53.4%) or month 6 (57.4%).
There were a total of nine insomnia-related TEAEs reported by 7 of 162 subjects (4.3%). The frequency of treatment-emergent insomnia was similar between subjects who received MTS (3.8%) and PTS (4.6%) in the antecedent study. All insomnia-related TEAEs were considered to be of mild or moderate intensity and none were serious or resulted in discontinuation from the study.
Effectiveness
Improved ADHD symptoms were reported in the ITT population for MTS treatment as demonstrated by a decrease in mean ADHD-RS-IV total scores from baseline of the antecedent study across all study visits (Fig. 6). The overall mean (SD) change in ADHD-RS-IV total score from baseline of the antecedent study to end point was −23.0 (10.77), p < 0.001.

Mean (standard error [SE]) Attention-Deficit/Hyperactivity Disorder–Rating Scale IV (ADHD-RS-IV) total scores by time point for antecedent methylphenidate transdermal system (MTS) and placebo transdermal system (PTS) treatment groups, intent-to-treat (ITT) population. End point is last nonmissing assessment obtained postbaseline. W = Week, M = month.
As early as the first postentry assessment, subjects who had previously received PTS in the antecedent study showed significant improvement in mean ADHD-RS-IV total scores in the current study (p < 0.005). In contrast, subjects who previously received MTS in the antecedent study exhibited significant worsening in mean ADHD-RS-IV total scores at week 1 (p < 0.001) and week 2 (p < 0.001) compared to entry into this study, probably as a result of having to reinitiate titration of MTS from the lowest dose. By week 4, statistically significant improvement from the current study entry was observed in the antecedent MTS group, and these observations continued through end of the study (p < 0.001). At entry in this study, more than three-quarters of the subjects (86/106, 81.1%) treated with MTS in the antecedent study were considered responders to MTS (>25% decrease in ADHD-RS-IV total score from baseline of the antecedent study). At end point in this study, the majority (93/106, 87.7%) of these subjects was considered responders to MTS using the same criteria.
Similar results were seen with the ADHD-RS-IV subscale scores; an analysis of ADHD-RS-IV hyperactivity/impulsivity and inattentiveness scores for all subjects in the ITT population showed significant improvement at end point relative to the mean subscale scores at baseline of the antecedent study (p < 0.001). The overall mean (SD) change in ADHD-RS-IV hyperactivity/impulsivity and inattentiveness scores from baseline of the antecedent study to end point were −9.9 (6.24) (p < 0.001) and −13.1 (6.28) (p < 0.001), respectively.
Starting at the first postentry week of the current study and continuing throughout the duration of the study, parents/guardians of subjects reported improvement in symptom amelioration as assessed by CPRS-R scores (p < 0.001). The overall mean (SD) change in CPRS-R total score from baseline of the antecedent study to end point was −27.6 (19.04). Subjects who had received PTS during the antecedent study showed significant improvement from baseline of the antecedent study after 1 week of treatment with MTS during the current study (p < 0.001).
CGI-I and PGA ratings were categorized as either “very much improved” or “much improved” in 75.9% (120/158) and 63.3% (100/158) of subjects at end point relative to baseline of the antecedent study, respectively. No differences in CGI-I responder rates were evident between the antecedent MTS (76.4%) and PTS (75.0%) groups at end point. The percentage of subjects with improvement on PGA scores was similar between the antecedent MTS (65.1%) and PTS (59.6%) groups at end point.
The self-reported Y-QoL-R total perceptual score significantly increased from baseline of the antecedent study to endpoint (p < 0.001) with MTS treatment and indicated an improvement in QoL parameters that are perceptual in nature. All four Y-QoL-R perceptual domain scores (self, relationship, environment, and general QoL) showed significant improvements from baseline of the antecedent study to end point (p ≤ 0.002). Overall, the majority of the Y-QoL-R contextual score items (i.e., parameters that describe the subject's life) did not show statistically significant improvement from baseline of the antecedent study at end point.
Discussion
This study shows that extended use of MTS given in doses of up to 30 mg/9 hours in adolescents with ADHD appears to be generally well tolerated and seems to provide sustained effectiveness throughout 6 months of dosing. The tolerability and effectiveness results in this study are consistent with those of previous studies of MTS in children and adolescents (McGough et al. 2006; Findling et al. 2008; Wilens et al. 2008; Findling et al. 2009), as well as in studies of oral ADHD medications (Wilens et al. 2003; McGough et al. 2005; Spencer et al. 2005; Kratochvil et al. 2006; Spencer et al. 2006; Wilens et al. 2006), with the exception of application-site reactions. The tolerability profile in the antecedent study (Findling et al. 2010) was comparable to that observed in the current study, in which most AEs were mild to moderate in severity. The most common TEAEs in the present study were decreased appetite (15.4%), headache (11.7%), and upper respiratory tract infection (10.5%). During the study there were a total of five SAEs reported; 3 (clonic convulsion, grand mal convulsion, and syncope) were considered not related to study treatment and 1 subject reported auditory and visual hallucinations that were considered related to treatment.
Previous reports indicate that generic instruments (i.e., an instrument not specified for use in a particular disease state) are capable of detecting differences in the QoL in children, adolescents, and adults with ADHD (Rentz et al. 2005; Varni and Burwinkle 2006; Adler et al. 2006). In this trial, the validated Y-QoL-R instrument was capable of detecting significant improvements in the total perceptual score; however, the Y-QoL-R was not able to detect statistically significant improvements in any items of the contextual module. Unlike other generic instruments used to assess QoL in subjects with ADHD that use proxy reports in children and adolescents (Rentz et al. 2005; Varni and Burwinkle 2006), the Y-QoL-R utilizes self-reports to evaluate QoL. Previous work has indicated differences in scores between proxy- and self-reporting of the same instrument (Theunissen et al. 1998), which may help to explain why differences were not evident for any of the contextual score items. Although further work in subject-reported outcomes should be completed in the adolescent population for the development of an appropriate tool to assess QoL in adolescents with ADHD, these results suggest that QoL may be improved with MTS treatment in adolescent subjects with ADHD.
Limitations
This study had several limitations. This study used an open-label design, thus the tolerability and effectiveness assessments are susceptible to observer bias. In addition, the lack of a placebo arm may confound the ability to precisely attribute the observed effectiveness to study treatment. Another way in which the results of long-term, open-label continuation studies may be biased is that subjects who enroll into a study after participation in an antecedent trial may only be that subset of patients who either have had improvements in ADHD symptoms and/or who do not experience substantial AEs in the antecedent study.
Subjects who failed to respond to psychostimulants in the past, those with conduct disorder, and those with other psychiatric co-morbidities were excluded from participating in this and the antecedent double-blind study. In addition, youths with substantive general medical conditions were also not enrolled into this clinical trial. Therefore, the results of this study should not be extrapolated to patient populations who were not included in this study.
In comparison to a 6-month, open-label trial of osmotic-release oral system (OROS) MPH in adolescents (Spencer et al. 2005), the attrition rate in this study was higher (46% vs. 29%). This is likely due to a high number of subjects who withdrew consent rather than discontinue from the study as the result of AEs or lack of efficacy. Moreover, nonadherence to study medication may be a contributing factor to attrition as studies have shown nonadherence is common, in both clinical trials and claim analysis studies, among subjects with ADHD receiving treatment (Adler and Nierenberg 2010).
Although a reduction in ADHD symptoms was observed by clinicians during the 6-month study of MTS in adolescents, no teacher-rated measures were used to ascertain effectiveness in an academic setting. Consequently, the results of this study should not be used to suggest that MTS improves academic performance in adolescents. Overall, the distribution of dermal scores (DRS and EODP) was generally similar across doses and visits. Higher maximum dermal scores occurred in subjects receiving 30 mg MTS compared to subjects receiving 10 mg, 15 mg, and 20 mg of MTS; however, across all MTS doses, no relationship between maximum dermal scores and subject disposition (study completion status) were apparent. This study was not designed or powered to determine the effect of each MTS dose on either safety or effectiveness measures. Further studies would be needed to assess the relationship between MTS dose and safety and effectiveness measures.
Conclusion
This 6-month, open-label extension study of MTS in adolescents with ADHD was completed by 54% of subjects. The most common reasons for early termination in this long-term study included withdrawal of consent (36.0%) and lost to follow up (25.3%); both are common reasons for discontinuation in such long-term studies. Overall, MTS was generally well tolerated, and AEs were generally typical of those associated with oral MPH, with the exception of application-site reactions associated with transdermal delivery of MPH. A total of 13 (8.0%) subjects discontinued due to AEs. Those AEs leading to discontinuation in 2 or more subjects included affect lability (2/162, 1.2%), irritability (2/162, 1.2%), and application site reactions (4/162, 2.5%). In this study, MTS was associated with benefit, as rated by clinicians, parents, and subjects, in the treatment of adolescents with ADHD.
Footnotes
Disclosures
Dr. Findling has received research support, acted as a consultant, and/or served on speakers' bureaus for Abbott, Addrenex, AstraZeneca, Biovail, Bristol-Myers Squibb, Forest, GlaxoSmithKline, KemPharm, Johnson & Johnson, Eli Lilly, Lundbeck, Neuropharm, Novartis, Organon, Otsuka, Pfizer, Sanofi-Aventis, Sepracor, Schering-Plough, Shire, Solvay, Supernus Pharmaceuticals, Validus, and Wyeth. Dr. Katic has received research support, acted as a consultant, and/or served on speakers' bureaus for Forest, GlaxoSmithKline, Lundbeck, Merck, Novartis, Sanofi-Aventis, Sepracor, Shire, Somerset, and Wyeth. Dr. Rubin has received research support, acted as a consultant, and/or served on speakers' bureaus for Abbott, Addrenex, Cephalon, Eli Lilly, Johnson, Johnson, McNeal, Novartis, Shire, and UCB Celltech. Dr. Moon received research support, acted as a consultant, and/or served on speakers' bureaus for Abbott, AstraZeneca, CNS Response, Eli Lilly, Johnson & Johnson, McNeil, Novartis, Pfizer, Sanofi-Aventis, Shire, Synosia Therapeutics, Takeda, and UCB Inc. Drs. Civil and Li are employees of Shire Development Inc. Dr. Civil is an employee of Shire Development, Inc. At the time of this study, Dr. Li was an employee of Shire Development, Inc.
Acknowledgments
The authors would like to thank the parents and teenagers who participated in the study. The authors also acknowledge Teresa A. Oblak, Ph.D., of The JB Ashtin Group, Inc., Plymouth, Michigan, for assistance in the preparation of this manuscript based on an author-approved outline and for implementing author-suggested revisions. This assistance was funded by Shire Development, Inc.
Dr. Li served as the statistician for the analysis of data.
The methylphenidate transdermal system (MTS) is manufactured by Noven Pharmaceuticals, Inc. This study was funded by Shire Development Inc., which was involved in the study design, conduct, and data analysis.