
Abstract
Objective:
This secondary analysis examined the efficacy and tolerability of atomoxetine (ATX) dosed once (QD) versus twice (BID) daily in 55 children aged 6–12 with attention-deficit/hyperactivity disorder (ADHD).
Methods:
The original 8-week trial was designed to assess the benefits of adding behavioral therapy to ATX. In it, all subjects were treated openly with ATX, with 50% randomly assigned to additional behavioral treatments. Every subject was started on QD dosing with a target dose of 1.2 mg/kg per day. A switch to BID dosing was allowed at study midpoint to improve tolerability and efficacy. Subjects not responding to ATX at midpoint were also given the option of 0.6 mg/kg dose increase. ADHD and oppositional defiant disorder (ODD) symptoms, global functioning, side effects, and classroom performance were measured weekly.
Results:
There were 22 subjects (40%) who switched to BID dosing at midpoint (mean dose = 1.56 mg/kg per day) with the other 33 remaining on QD dosing (mean dose = 1.33 mg/kg per day). The BID group did not display any improvement in parent-rated ODD symptoms during the first 4 weeks of the study on QD dosing, but there was a significant improvement seen after the addition of the second ATX dose (p < 0.05). However, BID dosing was not associated with differential rates of change for parent-rated ADHD symptoms or impairment, teacher ratings, or other measures of classroom functioning. BID dosing was associated with decreased rates of stomachaches (p < 0.05) but more persistent appetite loss than QD dosing. The degree of improvement observed during the first half of the study in ratings of global impairment and ODD but not ADHD symptoms predicted a switch to BID dosing at midpoint (p < 0.05).
Conclusions:
The addition of an afternoon dose of ATX was associated with improved control of ODD symptoms at home, with no change in school functioning.
Introduction
These findings have led to the recommendation that changes in dosing schedule can enhance the tolerability and efficacy of ATX (Weiss et al. 2006). However, no prior pediatric study has directly compared once (QD) versus twice daily dosing (BID) to formally evaluate this recommendation. Moreover, the one study showing that morning dosing outperformed evening dosing (Block et al. 2009) relied on parent ratings spanning a time period of up to 3 weeks per rating. Parental capacity to accurately recall symptom levels 2–3 weeks prior may be limited, especially for evaluating mild differences in efficacy between different dosing schedules of an active medication versus the more notable differences between an active medication and placebo.
The one study in adults that addressed this issue found that BID dosing led to greater benefit and reduced rates of nausea (Adler et al. 2006), contradicting the indirect inferences from the pediatric trials that QD dosing would be just as effective as BID dosing (Michelson et al. 2002). These conflicting findings demonstrate the need to directly examine the issue in children as findings from adults may not translate to younger subjects.
Our center recently completed an 8-week randomized trial comparing the relative efficacy of ATX + behavior therapy (BT) versus ATX alone for improving functioning at home and at school, in children with ADHD (Waxmonsky et al. 2010). It was found that the addition of BT to ATX led to sizable gains at home and mild incremental effects at school. After 4 weeks of once daily dosing with ATX, the protocol allowed study physicians to stay with once daily dosing or switch to twice daily dosing as well as to increase the dose from 1.2 to 1.8 mg/kg per day for subjects not demonstrating an adequate response. A switch to BID dosing was also allowed to address tolerability concerns. This design enabled a post hoc assessment of the effects of switching from QD to BID dosing of ATX for a subset of the subjects. Based on the existing literature, it was hypothesized that switching from once to twice a day dosing would improve efficacy at home but not school and improve tolerability throughout the day.
Methods
Subjects
All 55 subjects were children (44 boys and 11 girls) between the ages of 6–12 (M = 8.62, SD = 1.58) found to have ADHD using criteria specified in the Diagnostic and Statistical Manual of Mental Disorders, 4th edition, Text Revision (DSM-IV-TR) (American Psychiatric Association 2000). Diagnoses were based on ratings by parents and teachers on the disruptive behavior disorders rating scale, which assesses the DSM-IV-TR symptoms of ADHD, oppositional defiant disorder (ODD) and conduct disorder (CD) (Pelham et al. 1992) and parent report on the Diagnostic Interview Schedule for Children (NIMH-DISC Editorial Board 1999). Parental report of impairing ADHD, ODD, and CD symptoms were confirmed by direct interview with a masters level or higher clinician using the Disruptive Behavior Disorder Interview (Pelham 1998). If a subject was already taking ADHD medication other than ATX, it was stopped for at least 48 hours before screening (all discontinued medications were stimulants).
Exclusion criteria included (1) current or past history of seizures (not including benign febrile seizures); (2) other physical conditions that precluded administration of ATX (i.e., marked cardiac conduction delay, etc.); (3) documented trial of ATX of 3 weeks or more on at least 0.8 mg/kg per day or a documented inability to tolerate this dose; (4) serious forms of psychopathology other than ADHD, such as autism, bipolar disorder, schizophrenia, or any other psychopathology requiring urgent treatment with psychotropic medication; (5) a history of major depression requiring treatment (therapy or medication) or any past history of self harm or serious suicidal ideation; (6) an IQ of < 75; and (7) no evidence of ADHD-related impairment at school.
The mean clinical global impression severity score for ADHD was 4.3, placing subjects in the moderate range for impairment. The majority of subjects (85.5%) were found to have ADHD-combined type (see Table 1). In addition, 22 (40.0%) also met criteria for CD and another 23 (41.8%) met criteria for ODD, leaving 10 (18.2%) with non-co-morbid ADHD. The racial composition of the sample was 80.0% Caucasian, 10.9% African American, and 9.1% mixed race, and the ethnic composition was 5.5% Hispanic and 95.5% non-Hispanic, matching the demographics of Western New York. There were 21 (38.2%) subjects who had never been treated with stimulants.
Values in table under “Dosing group” columns are means (with standard deviations in parentheses) or percentages. Values in table under “Statistical comparison” column are ANOVA f-tests or χ 2 tests (with degrees of freedom in parentheses).
Full scale IQ estimated from the block design and vocabulary subtests of the Wechsler Intelligence Scale for Children, 3rd ed. (Wechsler 1991).
Achievement scores on the Wechsler Individual Achievement Tests, 2nd ed. (Psychological Corporation 2002).
Nakao and Treas Socioeconomic Status (Nakao and Treas 1994).
p < 0.10.
Average symptom rating on the DBD Rating Scale (Pelham et al. 1992), where 0 is “not at all,” 1 is “just a little,” 2 is “pretty much,” and 3 is “very much.”
ADHD = attention-deficit/hyperactivity disorder; ANOVA = analysis of variance; ODD = oppositional defiant disorder; CD = conduct disorder; DBD = Disruptive Behavior Disorders.
One subject from the original study was excluded because he entered the original trial on an ATX dose above 1.2 mg/kg per day, which is greater than the dose at which study physicians were given the option to adjust dosing schedule. Six additional subjects (10% of the total subject pool) had begun to use ATX before study entry, including 3 who had just been started on ATX by their pediatrician (<2 weeks of usage) and 3 who were taking a potentially subtherapeutic dose (<0.8 mg/kg per day) for 2–4 weeks before study entry. The average duration of use before study entry for these 6 subjects was ∼2.5 weeks.
These subjects (four in the QD group and two in the BID group) were included in this analysis because the efficacy of ATX had not been established in any of the cases. These subjects followed the titration protocol described below except that they entered the study on their current dose. When the analyses reported below were recomputed after excluding these six subjects, the results were substantively the same. The study was approved by the Children and Youth Institutional Review Board at the Women's and Children's Hospital of Buffalo. Informed consent that included a detailed discussion of the risks of treatment was obtained from legal guardians and written assent was obtained from children before data collection. Table 1 summarizes demographic and rating scale data for subjects as a function of treatment condition.
Of the 55 children, 7 (12.7%) discontinued the study before completion, which included 4 children (12.1%) receiving ATX once per day and 3 (13.6%) receiving it twice per day. The rate of early dropout did not differ by dosing schedule, χ 2 (1) = 0.03, p = 0.869. Of the seven who stopped early, four children discontinued because the parent believed that the medication was ineffective (one of these also quit in part due to nausea while on ATX), two children refused ongoing medication, and one discontinued due to parental concerns over increased emotional lability (more prone to cry but no expression of suicidal thoughts). One-way analysis of variances comparing subjects who stopped early with those who completed the study showed no significant differences on any of the 25 baseline comparisons, suggesting that those who discontinued the study early were not different from those who completed the study. All available data for subjects who stopped early were included in analyses using SAS mixed models (described below).
Design
The original study consisted of an 8-week open label trial of ATX with one-half of the subjects randomly assigned to receive ATX + BT and the remaining subjects randomly assigned to receive ATX alone (ATX). All data were collected between March 2007 and May 2008. The clinical trial began after completing a baseline assessment, followed by 8 weeks of administering ATX (including a 3-week titration period). BT for children in the ATX + BT condition began simultaneously with the start of medication. Except as noted, outcome measures were completed at the end of each week. Details of the BT intervention are provided in the original study article (Waxmonsky et al. 2010).
Medication
All medication was dosed openly in the original protocol as the primary goal of the study was to compare the differences between ATX + BT versus ATX alone. A weight-based dosing protocol similar to prior studies of ATX was used (Michelson et al. 2002; Weiss et al. 2005). ATX was administered as a single morning dose for all subjects over the first 3 weeks. All subjects started on 0.5 mg/kg (rounded to the nearest 5 mg dose) for 3 days, then 0.8 mg/kg for the next 4 days. However, 10% of subjects had already been on medication for ∼2.5 weeks before baseline. On day 8 (week 2), all subjects were increased to 1.2 mg/kg in the form of a single morning dose. Phone visits were completed by a study physician at weeks 1 (before increasing to 0.1.2 mg/kg per day) and 2 to assess drug tolerability. Subjects were brought into the study center after 3 weeks of ATX usage to assess tolerability and efficacy of the medication. Consistent with past studies of ATX, study physicians were allowed to add a 0.6 mg/kg booster dose up to a maximum daily dose of 1.8 mg/kg per day in week 4 based on the presence of persistently impairing symptoms (Michelson et al. 2002; Weiss et al. 2005). In total, 36% of subjects (20/55) had their dose increased to 1.8 mg/kg per day. Dose decreases below 1.2 mg/kg per day were not allowed. At the same visit, study physicians were allowed to switch to twice daily (BID) dosing to address tolerability or efficacy concerns. Forty percent of the sample (22/55) was switched to a BID schedule with the first dose in the morning and the second dose in the late afternoon or early evening. The main reason for switching to BID dosing was to address residual symptoms later in the day (100%); 27% of these subjects also switched to improve ATX tolerability. The mean dose was 15% higher in the BID group (1.56 mg/kg per day) than the QD group (1.33 mg/kg per day) [F(1, 53) = 8.21, p = 0.006]. The mean dose in both groups was above the standard therapeutic range for ATX of 0.8–1.2 mg/kg per day (Waxmonsky 2005). There was a difference in total daily dose between groups because 59% (13/22) of subjects switched to BID dosing were increased to the maximal allowed dose of 1.8 mg/kg per day versus 21% (7/33) of those remaining on QD dosing for the duration of the study. It was not required that subjects in the BID group receive two equal doses; however, the BID group received 55% (0.86 mg/kg) of their total daily dose in the morning and 45% (0.7 mg/kg) later in the day, nearly approximating a balanced split dosing scheduled. No changes in dose or dosing schedule were allowed after the completion of week 4.
Subjects receiving BT were equally distributed across the two dosing groups (see Table 1). Despite the fact that the groups were not randomly assigned to dosing schedule, there were no baseline differences between the two groups except for a trend in the BID dosing group having a higher SES (see Table 1).
Assessments
Classroom observation
Observations were conducted at baseline and endpoint using the Student Behavior Teacher Response Observation Code (Pelham et al. 2008a). They were completed by two research assistants who received 10 hours of training in the observation system, including coding at least four videotaped assessments. Observers watched children in their classrooms for 30 minutes during an academic activity and recorded each time the subject violated a classroom rule. The number of total classroom rule violations was used as the primary outcome measure for the study. Other work by our group has found that ADHD youth will exhibit an average of 10 rule violations per 30-minute observation period (Fabiano et al. in press), whereas children without ADHD will exhibit two or less (Pelham et al. 2008b). Reliability for this study was evaluated by having a second observer independently code ∼30% of the total observations. Interrater reliability was 0.89 and the mean difference between raters was not significant, t(42) = 0.62, p = 0.538. Observers were blind to medication dose and schedule, but the primary observer was aware if subjects were in the BT group or not.
Daily report card/individual target behavior evaluation
A daily report card (DRC) was developed for each child in the ATX + BT group at the start of the clinical trial. Teachers developed individual treatment goals based on the child's current classroom functioning and evaluated the child's performance on these treatment goals at least twice per day. Children were given feedback on their DRC performance, and results were linked to school and home-based rewards in an effort to promote desirable behaviors. The child's ability to meet these goals was summarized by computing the total percent of goals achieved each week. A form similar to the DRC, called the individual target behavior evaluation (ITBE), was developed for children not in behavioral treatment (ATX only) (Pelham et al. 2005a). Like the DRC, the ITBE listed behavioral goals specific to each child, and teachers evaluated children's performance on these goals throughout the day, and results were used to calculate the percentage of goals met per week. However, for the ITBE, teachers did not provide feedback to the child about their performance, nor did parents monitor the form or provide a positive or negative consequence to reinforce the child's performance. Thus, the DRC served as both a measure of and treatment for children's school behavior, whereas the ITBE served as a measure of but not a treatment for school behavior.
Rating scales
IOWA Connors
The IOWA Connors measures inattentive-impulsive-overactive and oppositional-defiant (OD) behaviors in children (Loney and Milich 1982; Pelham et al. 1989; Waschbusch and Willoughby 2008). Items on the IOWA are evaluated using Likert scales that range from 0 (“not at all”) to 3 (“very much”). The IOWA was completed by parents and teachers at the end of each week.
Impairment rating scale
The impairment rating scale is a 6 (for teachers) to 8 (for parents) item measure that uses visual-analog scales to evaluate the child's impairment level and need for treatment in developmentally important areas (Fabiano et al. 2006). For this study, only the overall impairment item was used. The scale is scored from 0 (no problem) to 6 (extreme problem).
Pittsburgh Side Effects Rating Scale
The Pittsburgh Side Effects Rating Scale (PSERS) measures adverse events commonly associated with stimulant medication (Pelham 1993, 2002) and has been used in multiple studies of ADHD (e.g., Pelham et al. 2005b; Waxmonsky et al. in press). For this study, the PSERS was modified to also assess adverse emotional events that have been reported with ATX, including suicidal statements. The resulting scale consisted of 13 items (for teachers) or 14 items (an additional sleep item for parents) rated using Likert Scales that ranged from 0 (“none”) to 3 (“severe”). The PSERS was completed at the end of each week by parents and teachers. For this study, we examined a total side effect score, computed by averaging across all items, as well as individual side effect items that have been associated with dosing schedule in prior studies: (1) dull, tired, listless; (2) headaches; (3) stomachaches; (4) crabby, irritable; and (5) loss of appetite. We also examined rates of clinically significant side effects within each group, defined as a rating of moderate (2) or severe (3).
Children's depression rating scale—revised
The children's depression rating scale- revised (CDRS-R) is a 17-item scale that measures Diagnostic and Statistical Manual of Mental Disorders, 4th edition (DSM-IV) (American Psychiatric Association 1994) symptoms of depression, including suicidal thoughts (Poznanski and Mokros 1995). Items are completed using 1–5 or 1–7 scales, with 1 designating no difficulty with that symptom. It is the mostly widely used measure of pediatric depression in clinical trials (March et al. 2004). The CDRS-R was completed in an interview format with parents and children evaluated separately. Results were combined by taking the maximum (most depressed) scores across informant on an item-by-item basis. In cases of marked discrepancy between informants, the score deemed most reliable was used. The complete CDRS-R was administered pretreatment and posttreatment with the suicide item (item no. 13) administered over the phone at the end of weeks 1 and 2 as well. The total score and the suicide item were used in analyses.
Statistical analyses
To compare treatment response in the QD dosing group versus the BID dosing group, data were analyzed using SAS for Windows (version 9.2) Proc Mixed with time/treatment (weeks 1&2 vs. 3&4 vs. 5&6 vs. 7&8) and dosing group (QD vs. BID) as factors. Consistent with the original study, we refer to the factor evaluating change as time/treatment because it reflects the fact that time and treatment were not disentangled in this study and participant's change may have resulted from either. In evaluating time/treatment, 2-week averages rather than weekly averages were used because they included more data points and thus provide a more robust estimate of behavior. An advantage of SAS Proc Mixed is that it uses advanced methods for handling missing data that allow subjects with partial data to be incorporated into the analyses. Significant interactions were followed up with simple effects tests and pairwise comparisons of time/treatment weeks to determine the nature of change over time/treatment. Results of the analyses, as well as means and standard deviations for each group, are summarized in Table 2 for outcome measures and Table 3 for side effect measures. To evaluate measures that were collected at two time points (pretreatment and posttreatment) rather than weekly (i.e., classroom observations, CDRS-R, and children's weight), we computed 2 (dosing group) × 2 (time/treatment: Pre vs. post) analyses. These results are reported in text. Although there were significant main effects of time/treatment for nearly every measure, these were not followed up because the focus of this study was on the impact of a change in dose and dosing schedule for subjects exhibiting a suboptimal initial response and because a previous report presented treatment effects in this sample (Waxmonsky et al. 2010).
Within each row and group, means with different subscripts differ in post hoc comparisons. Overall Impairment measured using the impairment rating scale (Fabiano et al. 2006).
p < 0.05.
p < 0.10.
D = dosing group (QD vs. BID); DRC = daily report card; inatt-impul-hyper = inattention-impulsivity-hyperactivity as measured by the IOWA Conners rating scale (Loney and Milich 1982; Pelham et al. 1989; Waschbusch and Willoughby 2008); ITBE = individual target behavior evaluation; T = time/treatment; T × D = time/treatment × dosing group interaction; ANOVA = analysis of variance.
Within each row and group, means with different subscripts differ in post hoc comparisons. Side effects measured using the Pittsburgh side effects rating scale (Pelham 1993).
p < 0.05.
p < 0.10.
NA = not applicable because interaction was not significant in the overall ANOVA; T = time/treatment; D = dosing group; T × D = time/treatment × dosing group interaction.
After the group comparisons, a series of logistic regressions were computed to evaluate whether group membership can be predicted from change in parent or teacher ratings of behavior, impairment, or side effects early in treatment. For behavior and impairment ratings, the predictors were change between baseline assessment (before the start of treatment) and the fourth week of treatment. For side effect ratings, predictors were change between the first week of treatment and the fourth week of treatment. All change scores were coded so that higher change scores indicate greater improvement or increased side effects. Group membership was coded as 0 (for the QD group) or 1 (for the BID group). To save space, only regressions that significantly predicted group membership are described.
Results
Group comparisons of treatment outcomes
Classroom observations
There was a significant main effect of time/treatment [F(1, 48) = 14.00, p = 0.0005] and a marginal effect of Dosing Group [F(1, 53) = 2.93, p = 0.0929], but these were qualified by a significant dosing group × time/treatment interaction [F(1, 48) = 4.10, p = 0.0485]. Simple effects tests showed that the number of classroom rule violations declined over treatment for the QD dose group [F(1, 48) = 21.60, p < 0.0001] but not for the BID dose group [F(1, 48) = 1.22, p = 0.2789]. Examination of means and standard deviations shows that the QD group improved between pretreatment and posttreatment (pretreatment: M = 9.64, SD = 7.27; posttreatment: M = 3.48, SD = 4.50) whereas the BID group did not (pretreatment: M = 10.41, SD = 6.66; posttreatment: M = 8.05, SD = 8.89).
Daily report card/individual target behavior evaluation
As shown in Table 2, there was a marginally significant dosing group × treatment interaction. Follow-up tests showed that the QD group significantly improved over time/treatment, especially in the middle of treatment (between weeks 3&4 and 5&6), but the BID group did not significantly improve over time/treatment when given ATX once or twice a day.
Teacher ratings
Overall impairment
As shown in Table 2, there was a significant dosing group × time/treatment interaction. Simple effect tests showed that the QD group improved over time/treatment, with significant decreases at the start (between weeks 1&2 and 3&4) and end (between weeks 5&6 and 7&8) of treatment. In contrast, the BID group did not significantly change over time/treatment although nonsignificant reductions in impairment were seen.
Inattentive-impulsive-overactive
As shown in Table 2, there was a significant dosing group × time/treatment interaction, which is illustrated in Figure 1. Follow-up tests showed that both groups improved over time/treatment. The QD group improved at the start of treatment (between weeks 1&2 and 3&4) and again in the middle of treatment (between weeks 3&4 and 5&6). The BID group improved at the start of treatment (between weeks 1&2 and 3&4) but did not change thereafter, although initial treatment gains persisted through to the end of the study.

Teacher ratings of inattentive-impulsive-overactive behavior as a function of dosing group and time/treatment. Twice daily dose was begun at week 4.
Oppositional-defiant
As shown in Table 2, there was a significant dosing group × time/treatment interaction. Follow-up tests showed that the QD group improved over time/treatment, with significant decreases at the start (between weeks 1&2 and 3&4) and middle (between weeks 3&4 and 5&6) of treatment, but the BID group did not change.
Parent ratings
Overall impairment
As shown in Table 2, there was a significant dosing group × time/treatment interaction, which is illustrated in Figure 2. Follow-up tests showed that both groups significantly improved over time/treatment. For the QD group, impairment decreased at the start (between weeks 1&2 and 3&4) and middle (between weeks 3&4 and 5&6) of treatment. For the BID group, impairment improved at the start of treatment (between weeks 1&2 and 3&4) and again between the middle and end of treatment (between weeks 3&4 and 7&8).

Parent ratings of overall impairment as a function of dosing group and time/treatment. Twice daily dose was begun at week 4.
Inattentive-impulsive-overactive
As shown in Table 2, there was a significant main effect of time/treatment but no significant effects involving dosing group.
Oppositional-defiant
As shown in Table 2, there was a significant dosing group × time/treatment interaction, which is illustrated in Figure 3. Follow-up tests showed that ODD symptoms changed significantly over time/treatment for both groups. For the QD group, ODD symptoms decreased at the start of treatment (between weeks 1&2 and 3&4) and remained stable thereafter. For the BID group, symptom ratings did not change during the first half of the study then significantly improved in the middle of treatment (between 3&4 and 5&6) and remained steady thereafter.
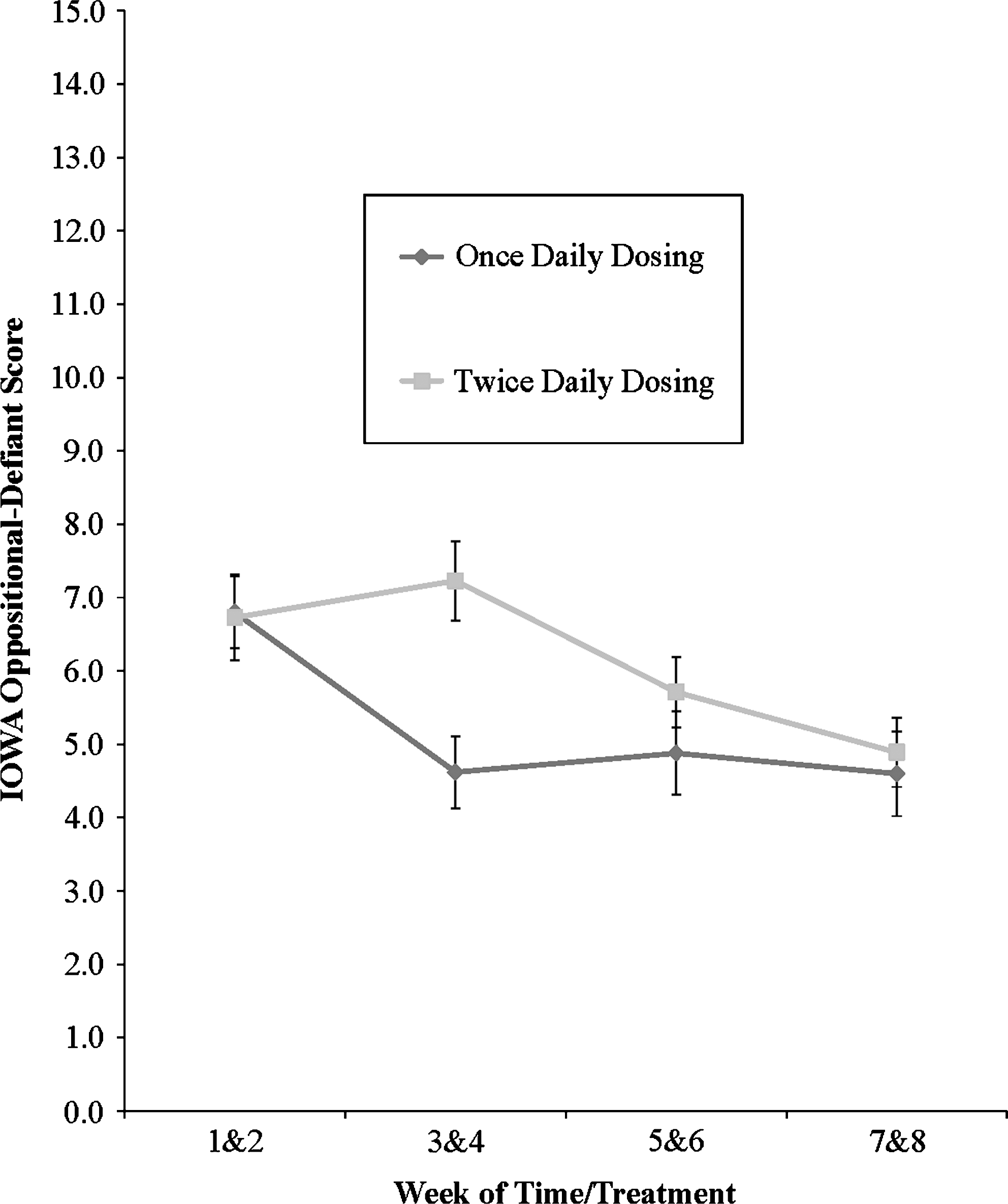
Parent ratings of oppositional-defiant behavior as a function of dosing group and time/treatment. Twice daily dose was begun at week 4.
Group comparisons of side effects
Teacher-rated side effects
Teacher side effect ratings were all in the mild range with no single item in either group having a mean rating higher than 0.63 at any assessment point (dull, tired, listless for BID group at weeks 3&4) on the 0–3 PSERS. The frequency of moderate or severe side effects (≥2 on the PSERS) was under 15% across all time points for both groups (highest rates were 13.6% for stomachaches and crabby/irritable in the BID group and 9% for headaches, stomachaches, and appetite loss in the QD group). As shown in Table 3, analysis of mean side effect ratings resulted in a marginally significant dosing group × time/treatment interaction for headaches and for stomachaches but not for other side effects (dull/tired/listless, crabby/irritable, appetite loss, and overall average). However, when analyzing headaches rated as only moderate or severe (vs. none or mild), the difference was no longer significant with rates gradually declining over time in both groups. The BID group had a marginally significantly greater rate of moderate to severe stomachaches at weeks 1&2 (0% for QD vs. 13.6% for BID, p < 0.10) but not thereafter. Follow-up tests showed that both groups changed over time. For the QD group, severity of stomachaches increased in the second half of the study, whereas for the BID group it decreased after the switch to twice daily dosing in week 4 and remained stable thereafter. At endpoint, 9% of the QD group reported moderate or severe stomachaches versus 0% in the BID group, although the difference did not reach significance.
Parent-rated side effects
Parents typically reported more side effects than teachers, although the weekly means were all in the mild range, with a high score of 1.18 for crabby/irritable behaviors in the BID group at weeks 1&2. The main effect of group for the overall average side effect score showed that parents in the QD group reported slightly less intense side effects in their child than did parents in the BID group (QD: M = 0.33, SD = 0.27; BID: M = 0.44, SD = 0.35). There were no significant group differences in the overall rate of moderate or severe side effects at baseline (61% for the QD group vs. 73% for the BID group) or endpoint (30% for QD vs. 41% for BID). As shown in Table 3, there was also a significant dosing group × time/treatment interaction for crabby/irritable and appetite loss but not for other individual measures. Follow-up tests for crabby/irritable showed that the QD group did not change over time but rates declined in the BID group (between weeks 5&6 and 7&8). Although the BID group had significantly higher rates of moderate or severe irritability at baseline (45.5% for BID vs. 18.2% for QD), the difference dissipated by endpoint (27.3% for BID vs. 9.1% for QD, p > 0.10). Before the midpoint dosage adjustments, the two groups had nearly identical rates of appetite loss. Follow-up tests showed change over time for the QD group, with improved appetite at the end of treatment (rates of moderate or severe appetite loss declined from 15.2% to 6.1%) but no change for the BID group (18.2% at baseline to 13.6% at endpoint). Endpoint ratings were not significantly different between the two groups.
Child weight
There was a marginally significant main effect of Dosing Group [F(1, 53) = 3.72, p = 0.0592], but the main effect of time/treatment and the dosing group × time/treatment interaction was not significant (p's > 0.71). The dosing group main effect showed that children in the QD group were marginally heavier than children in the BID group (QD: M = 36.02 kg, SD = 11.98; BID: M = 30.40 kg, SD = 9.98), but change in weight over the course of time/treatment did not differ between the groups (QD: Pretreatment M = 36.19 kg, SD = 12.34 and posttreatment M = 36.11 kg, SD = 11.78; BID: Pretreatment M = 30.05 kg, SD = 9.93 and posttreatment M = 30.16 kg, SD = 10.29).
Children's depression rating scale—revised
There were no significant main effects or interactions (p's > 0.30) involving dosing group for the CDRS-R total score (QD: Pretreatment M = 25.58, SD = 6.03 and posttreatment M = 23.61, SD = 6.80; BID: Pretreatment M = 24.23, SD = 3.89 and posttreatment M = 22.28, SD = 4.11). Likewise, there were no significant main effects or interactions (p's > 0.53) involving dosing group for the CDRS-R suicidal item (QD: Pretreatment M = 1.27, SD = 0.67 and posttreatment M = 1.13, SD = 0.52; BID: Pretreatment M = 1.36, SD = 0.73 and posttreatment M = 1.09, SD = 0.46). No new cases of suicidal ideation or mania were reported. Severity ratings of suicidal ideation (item 13 on the CDRS-R) increased temporarily for only one subject. Upon direct assessment of this subject, there was no evidence of suicidal ideation.
Logistic regressions predicting group membership
None of the regressions using teacher ratings was significant nor were regressions using the DRC/ITBE or classroom observations. However, three regressions using parent ratings were significant. First, change in parent-rated overall impairment predicted dose group [χ 2(1) = 4.67, p = 0.031]. The regression coefficient showed that less improvement in overall impairment during the first half of the study (weeks 1–4) was associated with membership in the BID group [B = −0.474, Wald = 3.72, odds ratio = 0.62, p = 0.054]. Second, change in parent-rated OD behavior predicted dose group [χ 2(1) = 4.45, p = 0.035]. The regression coefficient showed that less improvement in OD behavior was associated with membership in the BID group [B = −0.137, Wald = 3.93, odds ratio = 0.87, p = 0.048]. Finally, change in parent-rated loss of appetite was a marginally significant predictor of dose group [χ 2(1) = 3.68, p = 0.055]. The regression coefficient showed that less recovery of appetite by week 4 was associated with membership in the BID group [B = −0.738, Wald = 3.05, odds ratio = 0.48, p = 0.081].
Discussion
The primary goal of this analysis was to examine the impact of a change in dosing schedule on the efficacy and tolerability of ATX. In this study, midpoint ATX adjustments could include a change in dosing schedule (QD to BID), an adjustment in total daily dose (1.2–1.8 mg/kg per day) or both. Results showed that for the 22 subjects not demonstrating an adequate response to once daily dosing, adding an afternoon dose (either by spitting the morning dose or by adding additional medication later in the day) led to greater improvements at home with no changes seen at school. The original ATX study that served as the basis for this secondary analysis found greater effects of ATX when combined with BT than when ATX was administered alone, with the majority of differences occurring at home (Waxmonsky et al. 2010). These combined results provide clinicians with two options for enhancing the effects of ATX at home, namely, adding BT or an afternoon dose of ATX. Both options may be advisable in cases where there are severe, persisting symptoms at home. In addition, switching to BID dosing was associated with fewer stomachaches but no improvements in appetite.
The efficacy of ATX has been well established at both home and school (Michelson et al. 2001, 2002; Kelsey et al. 2004; Weiss et al. 2005; Waxmonsky et al. 2010). Comparable effect sizes from previous pediatric ATX studies using once versus twice daily dosing provided indirect evidence that the therapeutic effects of ATX for pediatric ADHD were similar regardless of dosing schedule (Michelson et al. 2001, 2002; Greenhill et al. 2007; Block et al. 2009). However, no work has formally examined the impact of dosing schedule on the degree of improvement in ADHD and ODD symptoms for pediatric populations. The one study in adults to address this issue found improved efficacy and tolerability with twice daily dosing (Adler et al. 2008). However, it is not clear that adult findings would apply to children, and the adult trial was powered to detect differential tolerability and not efficacy.
Improvement was observed in parent ratings of ODD within a week of making this dosing change (see Fig. 3). Previously, subjects in the BID group displayed no improvement in ODD symptoms while receiving a single morning dose of ATX. In contrast, the QD group improved steadily from weeks 0 to 4 and then leveled off, suggesting that the medication adjustments made at midpoint were associated with the additional improvement in ODD symptoms for the BID group. The incremental gains observed in the BID group after week 4 were sufficient to eliminate the significant group difference in parent-rated ODD symptoms that had developed by study midpoint (see Fig. 3). While no prior work in children has directly examined the impact of adjusting dosing schedule, past trials employing BID dosing found significant effects on ODD symptoms across a range of doses, whereas studies using a single morning dose have not consistently produced such effects (Newcorn et al. 2005; Biederman et al. 2007; Bangs et al. 2008). Moving the entire dose back to later in the day may accomplish the same effects but can lead to diminished daytime effectiveness (Block et al. 2009). Hence, adding an ATX dose later in the day may be the preferred pharmacological choice to address residual ODD symptoms at home in subjects not demonstrating adequate response to a single morning dose.
Despite an almost 30% reduction in the amount of medication given in the morning, a switch to BID dosing was not associated with any significant change in symptoms or impairment at school, even for the more objective measure of the DRC. In all cases, the week 8 teacher ratings for the BID group were nonsignificantly improved over the week 4 midpoint ratings. The only exception was the ADHD subscale on the IOWA Conners. Figure 1 shows that no further improvement in teacher-rated ADHD symptoms occurred after the midpoint medication adjustments in the BID group whereas the QD group showed continued reductions. However, the BID group had already declined to a very low level of teacher-rated ADHD symptoms by that time (mean rating of 1 or “mild” on the 5 item inattentive/overreactive subscale), suggesting that the subsequent plateau may have been due to a floor effect rather than the medication changes. Moreover, the endpoint ADHD ratings for the BID group were significantly improved over baseline and not different than the endpoint ratings for the QD group. Overall, there was little evidence that switching to BID dosing led to any appreciable change in symptom levels at school, presumably because subjects still received a morning dose that was within the standard therapeutic range for ATX (Waxmonsky 2005; Newcorn et al. 2009).
Unlike parent-rated ODD symptoms, there was no benefit of switching to BID dosing for parent-rated ADHD symptoms or impairment. For these latter two domains, the BID group had already evidenced significant improvements by study midpoint. Although there was steady improvement over the entire duration of the study, the improvement from weeks 4–8 was not greater than that seen during weeks 0–4. Therefore, it seems likely that ODD was specifically impacted because it was the only one of these three constructs that did not improve during the first half of the study in subjects later switched to BID dosing. Past studies have produced contrasting results as to whether youth with co-morbid ODD may exhibit diminished improvement in ADHD symptoms with ATX (Newcorn et al. 2005; Biederman et al. 2007; Bangs et al. 2008). The majority of our subjects showed an improvement in ADHD symptoms at home and school within the first few weeks of treatment with standard ATX dosing (1.2 mg/kg per day) even though 80% had co-morbid ODD/CD. These encouraging results for ODD may be due to the integration of BT and medication in this study as we have previously found that combined treatment leads to greater improvement in problem behaviors at home compared to medication alone (Waxmonsky et al. 2010). Together, these results support the early implementation of combined treatment strategies for children with ODD and ADHD who are prescribed ATX.
Adler et al. (2008) randomly assigned adults to once versus twice a day dosing and found 20% greater symptom improvements with BID dosing. Our study was the first to directly compare the effect of once versus twice daily dosing in children. Both of these studies support the efficacy of BID dosing schedules for ATX. The consistency of results across the lifespan is impressive given the vastly different demands made on school-aged children versus adults.
Although ATX was found to be generally safe and tolerable in this trial, there were two notable differences in the temporal pattern of side effects between the dosing groups. For subjects remaining on QD dosing, teachers reported more intense stomachaches over time although most were of mild severity. In contrast, teachers reported less intense stomachaches after the switch to a BID schedule despite the fact that the majority of subjects in the BID group had their dose increased at study midpoint (Table 3). This finding parallels what Adler et al. (2008) observed in adults taking twice a day ATX. Dividing the dose would lower the maximum blood concentration potentially improving tolerability as medication induced adverse events often occur at times of peak blood levels. Interestingly, parents reported less severe appetite suppression over time for the QD group, whereas no such decline was seen for the BID group despite comparable severity ratings through the first 4 weeks of the study in the two dosing groups. It could be that stomachaches and nausea relate mostly to peak blood levels, whereas appetite suppression is impacted more by duration of exposure within the day or total daily dose, which were both greater in the BID group. Hence, it may be advisable to switch to BID dosing when children report stomach upset in school but not for complaints of appetite loss without associated gastrointestinal discomfort.
Parent ratings of irritability declined after the switch to BID dosing, similar to what was found for parent ratings of ODD symptoms. This finding suggests that improved irritability ratings were more likely due to the enhanced efficacy of BID dosed ATX for ODD symptoms at home rather than to improved emotional tolerability of the drug when dosed twice a day. The lack of group differences in CDRS-R ratings also suggests comparable emotional tolerability between groups. There were no cases of new onset suicidal ideation in either group.
In a secondary analysis comparing adverse event rates between pediatric ATX studies using once versus twice daily dosing, Greenhill et al. (2007) also observed associations between dosing strategy and tolerability. Higher rates of adverse events with QD dosing were seen during the first 2 weeks on ATX, but by week 8 there were more reported side effects with BID dosing. We also found higher overall adverse events rates with twice daily dosing although this finding must be viewed with caution as difficulties tolerating the medication were one of the criteria for switching to BID dosing. Our study builds upon these prior results by specifically assessing the impact of a change in a dosing schedule on persistent side effects. Moreover, in the earlier study adverse events were measured by spontaneous report, whereas we employed a structured side effect scale that was administered to both parents and teachers.
While the decision to switch to twice daily dosing was supposed to be based on persistent ADHD symptoms, parent-rated impairment and ODD symptoms were a better predictor of dose schedule than were ADHD symptoms. These results are consistent with prior work showing that parents are more distressed by their children's oppositional and aggressive behaviors than their ADHD symptoms (Johnston and Mash 2001; Wymbs et al. 2008). The goal of our original ATX study was to examine the effect of adding BT to ATX, so parents enrolling in the study may have been seeking particular help with behavioral problems at home versus difficulties with inattention or hyperactivity-impulsivity. This interpretation is supported by the fact that nearly 80% of subjects had ODD or CD. The only other predictor of switching to BID dosing was appetite loss, which was actually less likely to improve after switching to twice daily dosing.
As is the case in any post hoc analysis, this study is subject to certain inherent limitations as it was not originally designed to address the impact of dosing schedule. Due to the lack of random assignment, the differences between the groups may be due to other uncontrolled factors rather than the difference in dosing strategy. Most notably, subjects were selected for the twice daily dosing group based on the presence of residual symptoms or difficulties tolerating the medication, so it was expected that those given BID dosing would show less improvement and report more intense side effects than those maintained on once a day dosing, especially before the midpoint dosing adjustments were made. Therefore, any comparisons between the two dosing groups should be interpreted with caution, especially for measures obtained only at baseline and endpoint, such as the classroom observations. However, the primary focus of this study was the within group effects of changing dosing schedule for the BID group. In addition, the groups were nearly identical on the numerous demographic and behavioral measures collected at baseline (see Table 1), making it less likely that there were other potential confounders impacting the between group comparisons.
Another limitation is that the statistical power to find group differences may have been limited because the analyses were not specified in the original study protocol. This issue is particularly relevant given the BID dose group consisted of only 22 subjects. However, this concern is lessened by the fact that the dosing schedule comparisons included a within subjects component, thereby increasing the power to detect changes due to dose schedule. Nonetheless, replication of this study using a larger sample would be beneficial. A third limitation is that the study lacked a control condition. Therefore, it is possible that parent ratings changed not because of the switch to twice daily dosing but because parents were aware that medication was being adjusted in an attempt to get a better treatment effect at home. However, placebo effects in clinical trials of ADHD are relatively small compared to other psychiatric conditions such as depression and as compared to the pharmacological effects of active ADHD medications (Waschbusch et al. 2009). The degree of change in ODD symptoms for ADHD youth administered placebo treatments in ATX studies is also comparably small (Newcorn et al. 2005; Biederman et al. 2007; Bangs et al. 2008).
Similar to the Greenhill analyses (Greenhill et al. 2007), dosing schedule (QD to BID) and total daily dose (1.2–1.8 mg/kg per day) were adjusted at the same time for some subjects. Most importantly, more subjects in the BID group had their dose increased, leading to a mean daily dose that was 15% higher than for the QD group. Therefore, it is unknown if the improvements in efficacy and tolerability seen in the second half of the study were due to the addition of the 0.6 mg/kg booster dose, the switch to BID dosing or simply to being on ATX for a longer time period. Newcorn et al. (2005) found that ATX-dosed BID leads to improvements in parent-rated ODD symptoms at doses as low as 0.5 mg/kg per day. In contrast, studies employing QD schedules at doses up to 1.8 mg/kg per day have not consistently found significant reductions in ODD symptoms over placebo (Biederman et al. 2007; Bangs et al. 2008), with no evidence of incremental effects for ATX doses above 1.25 mg/kg per day. A recent meta-analysis by Biederman et al. (2007) on the impact of co-morbid ODD on the response to ATX concluded that BID dosing may lead to a better control of ODD symptoms at home. These combined findings suggest that the majority of improvement in ODD symptoms occurs between 0.5 and 1.25 mg/kg. Similar results have been found for ADHD symptoms (Michelson et al. 2001; Kratochvil et al. 2007; Bangs et al. 2008) although one study found that doses over 1.2 mg/kg per day did improve ADHD symptoms but only for children with co-morbid ODD (Newcorn et al. 2005). All subjects were already at the high end of this dose range by the end of week 1 with no improvement seen in the BID group after 3 weeks on this dose when administered once per day. It is also possible that the BID group simply exhibited a delayed response to ATX that would have occurred after week 4 regardless of any change in dose or dosing schedule. However, response by week 4 predicts subsequent response in ATX trials where dosing schedule is held constant (Newcorn et al. 2009), so delayed responses to ATX are uncommon in subjects showing little improvement during the first month of treatment. Together, these findings suggest that the improvements in parent-rated ODD symptoms seen after week 4 in the BID group were due more to changes in dosing schedule than total daily dose or dosing duration. However, controlled trials that separate changes in dosing schedule from changes in total daily dose are needed before definitive conclusions can be drawn.
In regard to the tolerability findings, we focused on differential rates of change within the dosing groups rather than on between group differences in side effect severity. This choice was made because the BID group was partially comprised of subjects having a difficult time initially tolerating the medication, so that group would be expected to have more severe side effects as was seen for teacher-rated stomachaches.
The use of a structured side effect scale completed weekly by teachers and parents comprises a more detailed tolerability assessment of ATX than has been done in previous trials. However, it may have led to the detection of statistically significant differences with limited clinical relevance, as most side effect ratings were well within the mild range. For example, whereas the degree of change over time in teacher reported headaches was different between groups, 90% of all headaches were rated as mild. Nonetheless, parent-rated side effects predicted a change in dosing schedule, suggesting that even relatively mild side effects can impact prescribing patterns. The addition of pharmacokinetic data would have strengthened the findings, especially for the side effect results since it has been proposed that BID dosing may improve ATX tolerability by lowering C max (Greenhill et al. 2007).
The inclusion of six subjects who were started on ATX before entry (mean duration of prior use was 2.5 weeks) could have affected baseline ratings. In most cases, community pediatricians had started these subjects on ATX between referring them to the study and their initial intake assessment. They were included because the efficacy of ATX had not been established in any of these cases. Removal of these six subjects from the analyses did not substantively alter any of the findings. That the exclusion of subjects with extended durations of ATX usage did not alter the results reinforces the notion that changes in parent-rated ODD symptoms seen after week 4 were not due to a delayed response to ATX in the BID group. Lastly, the subjects were mostly Caucasian boys, which may limit the generalizability of the results.
Conclusions
The results of this study indicate that the addition of an afternoon dose of ATX, either by dividing the morning dose in two or by adding a second dose, may lead to a reduction in oppositional behaviors at home. A change in dosing schedule did not impact teacher-rated symptoms or classroom functioning. The parent trial for this secondary analysis demonstrated the utility of adding BT to ATX when there is a need for greater symptom control at home. The combined results provide clinicians with two alternatives to switching medication for children prescribed ATX who continue to exhibit impairing levels of ODD symptoms at home: Adding BT or a second dose of ATX later in the day. Children switched to BID dosing reported less stomachaches despite an increase in total daily dose. However, moving to BID dosing was associated with more persistent appetite loss. Hence, different dosing schedules may have their own unique tolerability profile. Further comparisons of once versus twice a day dosing under controlled settings are warranted based on these results.
Footnotes
Disclosures
The original dataset for this secondary analysis came from an investigator initiated study funded solely by Eli Lilly. No study personnel received compensation from Eli Lilly for this secondary analysis, other consulting projects, or any other services beyond the scope of conducting the original project. In the past 3 years, Dr. Waxmonsky has served on the Speaker's Bureau for Novartis and received honoraria from Scepter (CME Company) and is receiving research support from Shire Pharmaceuticals. Dr. Waschbusch and Pelham are also receiving research support from Shire Pharmaceuticals. In addition, Dr. Pelham has received honoraria from Janssen and holds common stock in Abbott Laboratories. Since submission of this article, Drs. Waxmonsky, Waschbusch, and Pelham have moved to Florida International University. Dr. Akinnusi has no disclosures to report.
This study was an investigator initiated trial funded entirely by Eli Lilly, Indianapolis, IN. Dr. Waschbusch served as the statistical consultant.