
Abstract
Objective:
This preliminary, 32-week study assessed the safety, tolerability, and pharmacokinetics of duloxetine in pediatric patients (aged 7–17 years) with major depressive disorder.
Methods:
Patients received flexible duloxetine doses of 20–120 mg once daily, with dose changes made based on clinical improvement and tolerability. Pharmacokinetic samples were collected across all duloxetine doses, and data were analyzed using population modeling. Primary outcome measures included treatment-emergent adverse events (TEAEs), vital signs, and Columbia-Suicide Severity Rating Scale (C-SSRS).
Results:
Of the 72 enrolled patients, 48 (66.7%) completed acute treatment (18 weeks) and 42 (58.3%) completed extended treatment. Most patients (55/72; 76%) required doses ≥60 mg once daily to optimize efficacy based on investigator judgment and Clinical Global Impressions-Severity score. Body weight and age did not significantly affect duloxetine pharmacokinetic parameters. Typical duloxetine clearance in pediatric patients was ∼42%–60% higher than that in adults. Four patients (5.6%) discontinued due to TEAEs. Many (36/72, 50%) patients experienced potentially clinically significant (PCS) elevations in blood pressure, with most cases (21/36, 58%) being transient. As assessed via C-SSRS, one nonfatal suicidal attempt occurred, two patients (2.8%) experienced worsening of suicidal ideation, and among the 19 patients reporting suicidal ideation at baseline, 17 (90%) reported improvement in suicidal ideation.
Conclusion:
Results suggested that pediatric patients generally tolerated duloxetine doses of 30 to 120 mg once daily, although transient PCS elevations in blood pressure were observed in many patients. Pharmacokinetic results suggested that adjustment of total daily dose based on body weight or age is not warranted for pediatric patients and different total daily doses may not be warranted for pediatric patients relative to adults.
Introduction
Duloxetine, a dual serotonin and norepinephrine reuptake inhibitor, has a 3-fold greater potency at the serotonin reuptake transporter than at the norepinephrine reuptake transporter using in vitro human cell lines. (Bymaster et al. 2005) It was first approved in 2004 for treating adults with MDD and has not been systematically evaluated for use in pediatric patients with MDD or any other indication.
The safety of duloxetine has been investigated in healthy adult volunteers at doses up to 400 mg/day (Derby et al. 2007) and in adult patients with MDD at doses up to 120 mg/day. (Hudson et al. 2005) Duloxetine pharmacokinetics have been evaluated in healthy adult volunteers at doses up to 120 mg/day (Skinner et al. 2003; Lobo et al. 2008) and in adult patients with hepatic (Suri et al. 2005) and renal impairment. (Lobo et al. 2010) Duloxetine is mainly eliminated through hepatic metabolism, predominantly by cytochrome P450 1A2 and, to a lesser extent, by the 2D6 isoenzyme. (Skinner et al. 2003; Lobo et al. 2008) The circulating metabolites are inactive. In adult patients, gender, smoking status, age, dose, and ethnic origin were identified as statistically significant covariates that influenced duloxetine pharmacokinetic parameters; however, based on the magnitude of change and inter-patient variability, specific dosing recommendations regarding these factors are not necessary. (Lobo et al. 2009).
This preliminary study, the first clinical study of duloxetine in patients <18 years of age, was designed to evaluate the safety, pharmacokinetics, and range of tolerable doses in a small group of children and adolescents (age 7–17 years) diagnosed with MDD. The dose range of 20 to 120 mg/day was selected to allow an extensive exploration of safety, tolerability, and pharmacokinetics of duloxetine across a wide dose range in pediatric patients of differing ages, gender, body weights, and sexual maturation. Results from this study will inform the design and dosing regimen for subsequent efficacy trials.
Methods
This study was approved by the ethics review boards for each study site. According to the principles of the Declaration of Helsinki, a parent/legal representative of each study patient provided written informed consent, and patients provided written assent prior to administration of any study drug or study procedures. Patients participated in the study at 22 sites in the United States from August 2007 to September 2008.
Study population
Study participants were outpatient children (age 7–11 years) and adolescents (age 12–17 years), who met Diagnostic and Statistical Manual of Mental Disorders, 4th edition Text Revision (DSM-IV-TR)(American Psychiatric Association 2000) criteria for MDD, and had a Children's Depression Rating Scale–Revised (CDRS-R)(Poznanski et al. 1984) total score ≥40 and a Clinical Global Impressions-Severity (CGI-Severity)(Guy 1976) score ≥4 at the three screening visits. An MDD diagnosis was supported by the Kiddie Schedule for Affective Disorders and Schizophrenia for School-Age Children Present and Lifetime Version (K-SADS-PL) (Kaufman et al. 1997) and was conducted by two independent evaluators, with at least one evaluator being a psychiatrist. Patients were also required to have normal or clinically unremarkable results on the physical examination, laboratory tests, and electrocardiogram completed at the screening visits. Female patients were required to have a negative serum pregnancy test at screening. Patients were excluded from the study if they were pregnant or lactating, had a <20 kg baseline weight; current and primary Axis I disorder (other than MDD) requiring pharmacotherapy; first-degree relative with bipolar disorder; serious or unstable medical illness; serious risk of suicide; history of substance abuse/dependence within the last year; or an unexplained positive urine drug screen. Generally, use of concomitant medications having primarily central nervous system activity was not permitted during the study.
Measures
Study drug compliance at each visit was defined as taking between 80% and 120% of the required number of capsules prescribed over a given time period and was assessed by patient/caregiver report and pill count. The Columbia-Suicide Severity Rating Scale (C-SSRS)(Posner 2007) was collected at baseline and every subsequent study visit to prospectively assess and then monitor changes in suicidal ideation and behaviors. Spontaneously reported treatment-emergent adverse events (TEAEs), weight, sitting blood pressure, and heart rate were collected at every study visit. Blood for chemistry and hematology laboratories was collected at baseline and at weeks 4, 10, 14, 18, 30, and early termination. Additional collections for chemistry occurred at weeks 2 and 6. Electrocardiograms were recorded at baseline and weeks 10, 18, 30, and early termination. Blood samples for CYP2D6 genotyping were collected at baseline (note there is no available method for CYP1A2 genotyping). One blood sample per visit for pharmacokinetic analysis was collected at weeks 2, 4, 6, 8, 10, 14, 18, and early termination. The clinician-rated CDRS-R (Poznanski et al. 1984) and CGI-Severity (Guy 1976) were completed at every visit.
Study design
This was a 32-week, phase 2, single-arm, flexible-dose, open-label study. The primary objective was to assess the safety and tolerability of duloxetine in the treatment of children and adolescents with MDD. The study consisted of five study periods: 2- to 4-week screening phase, 10-week dose titration/pharmacokinetic sampling phase with weekly study visits, 8-week acute safety and tolerability phase with visits every 2 weeks, 12-week extended safety and tolerability with visits every 4 weeks, and a 2-week taper phase.
Depending on body weight, patients meeting entry criteria initiated duloxetine treatment at 20 mg once daily (patients ≤40 kg) or 30 mg once daily (patients >40 kg) for 2 weeks, followed by flexible dose escalation ranging from 30 to 120 mg once daily over the remaining 8 weeks of the dose titration phase. Patients were required to remain at the initial duloxetine dose (20 or 30 mg once daily) for 2 weeks, and were then allowed to incrementally increase the daily dose from 20 to 30 to 60 to 90 to 120 mg at any of the eight remaining weekly study visits during the 10-week dose titration period. Dose escalations were based on the investigator's assessment of safety and tolerability and inadequate clinical response (CGI-Severity score ≥3). Dose decreases due to lack of tolerability were allowed provided they were not reduced below initiation dose levels. Patients who did not achieve a minimum level of clinical benefit (CGI-Improvement score ≤3) at the end of the dose titration phase were discontinued from the study. Patients continuing in the study entered the 8-week safety and tolerability phase during which they remained on the dose they attained during the dose titration period. During the subsequent 12-week extended safety and tolerability phase, flexible-dose titration within the range of 20 to 120 mg once daily was allowed at the investigator's discretion. A 2-week tapering phase allowed a gradual dose decrease to minimize discontinuation symptoms.
The duloxetine doses used in this study were 20, 30, 60, 90, and 120 mg once daily. This dose range was based on the dose range systematically evaluated in adults. The lowest doses used in the current study (20 and 30 mg once daily) are 50% of the recommended daily doses for adults with MDD. The maximum dose of 120 mg once daily was the highest dose systematically evaluated in adult clinical trials of duloxetine for MDD. (Hudson et al. 2005)
Data analysis
No formal statistical analysis was performed to determine the sample size for this study. To have at least four completers per age stratum of 7 through 9 years, 10 through 12 years, 13 through 14 years, and 15 through 17 years, ∼64 patients were required to be enrolled, assuming a drop-out rate of 75%. Further, tolerability and safety data from 64 patients would provide reasonable information to support dose selection for future duloxetine studies in pediatric patients.
Effectiveness and safety analyses were conducted on an intent-to-treat basis. A priori specified analyses of safety and tolerability measures were performed for the combined 10-week dose titration and 8-week safety and tolerability phases (collectively referred to as the acute treatment phase), and the 12-week extended safety and tolerability period (referred to as the extension phase).
Last-observation-carried-forward imputation was the prespecified primary methodology to evaluate mean changes in safety and effectiveness data. When last-observation-carried-forward mean change from baseline to end point was assessed, only patients with a baseline and a postbaseline measure were included in the analysis. Baseline was defined as the latest nonmissing observation across all three visits in the screening phase before active treatment began. End points for the acute treatment phase and extension phase were defined as the last nonmissing observation during the relevant phases.
Modal dose, serious adverse events (SAEs), discontinuations due to adverse events, and TEAEs (events that first occurred or worsened during the specified treatment phase) were summarized. Potentially clinically significant (PCS) changes were defined as the following changes from baseline: ≥3.5% decrease in weight; ≥5 mmHg increase in blood pressure to a value above the 95th percentile based on gender, age, and height; (National High Blood Pressure Education Program Working Group 2004) ≥20 bpm decrease in heart rate to a value no greater than 65; ≥25 bpm increase in heart rate to a value no less than 110. Abnormal laboratory results were based on reference ranges provided by Covance Central Laboratory Services (Covance Inc., Princeton, NJ).
Data from the C-SSRS were summarized for each category of suicidal behavior and suicidal ideation for both the acute treatment phase and the extension phase. A patient had worsening of suicidal ideation if the maximum suicidal ideation category during treatment was greater than the maximum suicidal ideation category during baseline. A patient had improvement in suicidal ideation at end point if the end point suicidal ideation category was less than the maximum suicidal ideation category during baseline.
For the CDRS-R a mixed-effects model repeated measures (MMRM) analysis was prespecified. The MMRM model included visit and baseline value terms. For longitudinal mean changes on the CDRS-R, MMRM results and observed case (OC) results are presented. For OC analyses of longitudinal mean changes, all data available were used with no imputation performed.
Two post hoc analyses of study safety and effectiveness data were completed: Sustained elevation in blood pressure (defined as three consecutive observations of PCS systolic or diastolic blood pressure) and remission rate (defined as a CDRS-R total score of ≤28) at end point during the acute treatment phase.
No statistical comparisons between subgroups were prespecified for any outcome.
Pharmacokinetic analysis
Collection of seven pharmacokinetic samples from each patient was planned during the 18-week acute phase of the study. Actual dosing of study drug (e.g., whether a patient missed a dose or took two doses at the same time) was assessed through analysis of the patient/caregiver report and pill count. Dose history (time and amount) for the last three doses prior to collecting the blood sample was taken into account in the pharmacokinetic analysis. Plasma concentrations of duloxetine were determined using a previously described, (Skinner et al. 2003) validated, high-performance liquid chromatography with tandem mass spectrometry (LC/MS/MS) method at Prevalere Life Sciences, Inc. (Whitesboro, NY). The time course of duloxetine plasma concentrations after oral administration of duloxetine was characterized using population pharmacokinetic modeling method of analysis. The analysis was conducted using nonlinear mixed-effects modeling program (NONMEM, Version VI, ICON Development Solutions, Ellicott City, MD). The first order conditional estimation method with interaction estimation was used. (Beal and Sheiner 1992) The modeling methodology included a stepwise approach of developing a base model, determining potential factors that may influence duloxetine pharmacokinetics (such as body weight, age, gender, ethnic origin, dose, CYP2D6 status, creatinine clearance, and menarche status) and evaluating the final model. The pharmacokinetic parameters of duloxetine in pediatric patients were compared with those reported in adult patients. (Lobo et al. 2009) Additionally, model-predicted duloxetine concentration-time profiles at steady state in pediatric patients and adults were compared using Monte Carlo simulation in 5,000 adult and pediatric patients using the final base model parameter estimates, between-subject variability, and residual error.
Results
Patient characteristics
All 72 enrolled patients took at least one dose of study drug and provided postbaseline data. Fifty-eight patients (80.6%) completed the 10-week dose titration phase, 48 patients (66.7%) completed 18 weeks of acute treatment, and 42 patients (58.3%) completed an additional 12 weeks of extended treatment (Fig. 1). Specifically by age group, the number of patients completing the acute treatment phase was 16 for 7 to 9 year olds, 15 for 10 to 12 year olds, 12 for 13 to 14 year olds, and 12 for 15 to 17 year olds. At least 85% of patients were compliant with study drug at each visit.
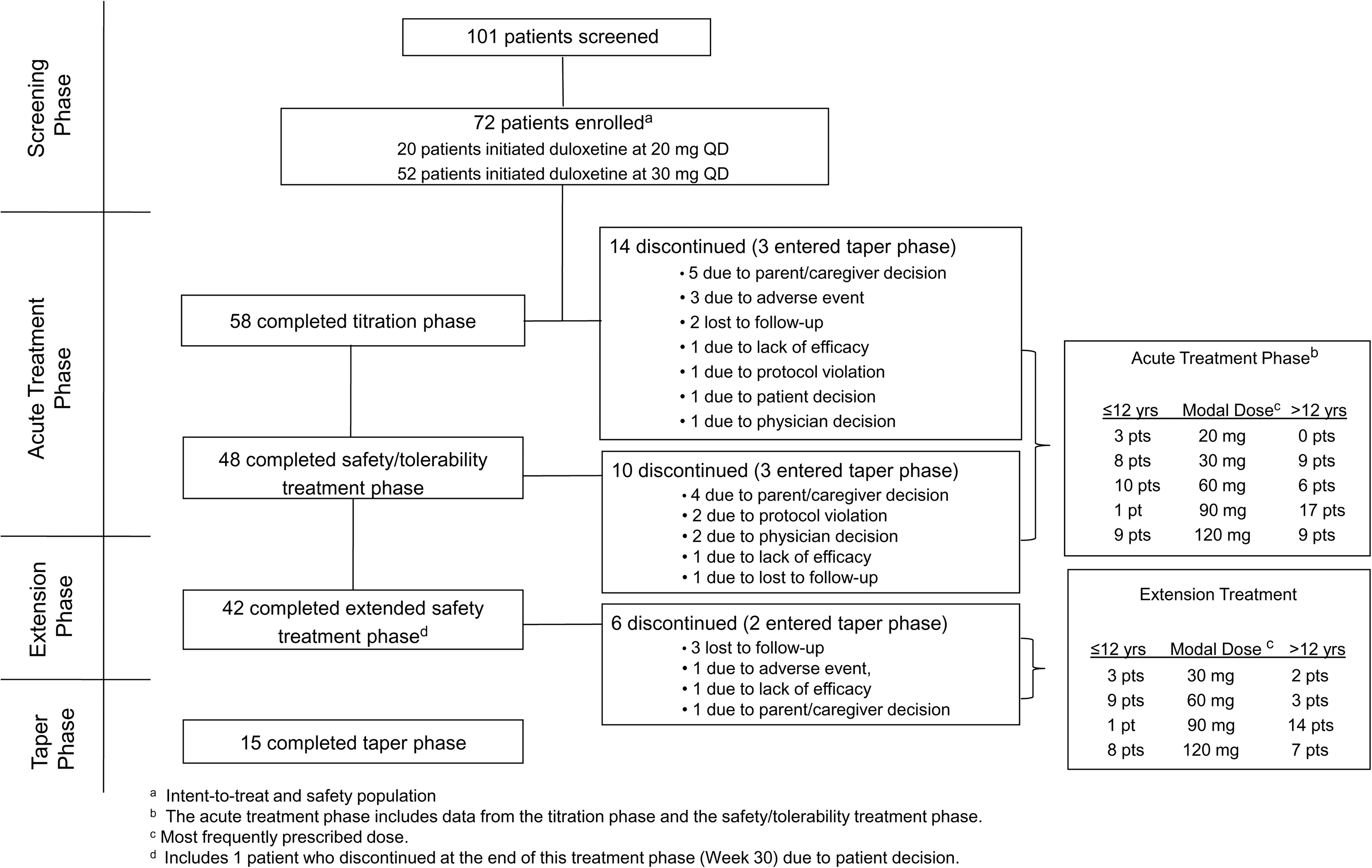
Consort diagram for Study HMFN. QD=once daily; pts=patients.
Baseline patient demographics and psychiatric profiles are shown in Table 1. The majority of study patients were experiencing their first MDD episode (median number of previous episodes was 0), and the average age of first episode was ∼11 years. Concomitant medications used by ≥5% of patients during acute treatment were ibuprofen (11.1%), paracetamol (9.7%), and salbutamol (5.6%). During the extension phase, the only concomitant medication reported with a frequency >5% was ibuprofen (6.3%). The majority of patients were identified as CYP2D6 extensive metabolizers (EMs) with four patients (5.6%) identified as CYP2D6 poor metabolizers (PMs).
Sixty-three percent of female patients had attained menarche.
Based on a total of 68 patients for whom data were obtained.
N=total number of patients; SD=standard deviation; BMI=body mass index; n=number of patients in the indicated category; CGI-Severity=Clinical Global Impressions-Severity; CDRS-R=Children's Depression Rating Scale-Revised.
Dosing
The majority of patients (55/72; 76%) required escalation to higher duloxetine doses (60, 90, or 120 mg once daily) during the 10-week dose titration period to optimize effectiveness. Similarly, most patients (52/72, 72%) had modal doses of 60, 90, or 120 mg once daily during the acute treatment phase, and 87% (42/48) of patients had modal doses of 60, 90, or 120 mg once daily during the extension phase (Fig. 1).
Safety
No deaths were reported during this study. Five patients (6.9%), four of whom were female, reported six SAEs (requiring hospitalization) during the study, with all SAEs beginning during the acute treatment period. One patient (60 mg) exhibited nonsuicidal self-injurious behavior (cutting), and one patient (90 mg) exhibited self-injurious behavior (choking considered an actual attempt) and experienced suicidal ideation. The third patient, after abruptly stopping duloxetine (90 mg), experienced worsening of depression. The other SAEs, viral gastroenteritis (60 mg) and worsening of oppositional defiant disorder (30 mg), were reported by one patient each. Of the five patients who experienced SAEs, two completed 30-weeks of treatment (patients with viral gastroenteritis and self-injurious behavior–cutting). The patient with self-injurious behavior (choking) discontinued because of lack of efficacy, the patient with oppositional defiant disorder discontinued because excluded medications were required to manage oppositional defiant disorder, and the patient with worsening of depression was lost to follow-up despite multiple attempts to contact.
Four patients (5.6%), two of whom were males, discontinued because of a TEAE. During the acute treatment period, three of the four patients discontinued while on duloxetine 30 mg once daily because of either nausea/vomiting, rash, or re-emergence of attention-deficit/hyperactivity disorder. The fourth patient, on duloxetine 120 mg once daily, discontinued because of irritability after ∼5 months of treatment.
The TEAEs occurring with a frequency ≥5% during acute treatment are listed in Table 2. Patients were more likely to experience a TEAE during the acute treatment period (57/72; 79.2%) than during the extension safety period (21/48; 43.8%). Nausea was the most frequently reported TEAE overall (25%). Females experienced nausea (14/35; 40.0%), vomiting (6/35; 17.1%), and dizziness (5/35; 14.3%) during acute treatment more frequently than males (4/37 [10.8%], 4/37 [10.8%], 2/37 [5.4%], respectively). With respect to other TEAEs of interest in the pediatric population, no events of mania or hypomania were reported. One patient experienced flight of ideas and agitation (1/72; 1.4%) and two patients experienced irritability (2/72; 2.8%) during acute treatment, with no patient experiencing flight of ideas or agitation and one patient experiencing irritability (1/48; 2.1%) during extension treatment.
Adverse events reported during the acute treatment period in at least 5% of patients.
The TEAEs reported during extension treatment (Week 18 through Week 30).
N=total number of enrolled patients; n=number of patients with specified treatment-emergent adverse event.
As assessed by the C-SSRS, two patients (2.8%) reported worsening of suicidal ideation from baseline (one during acute treatment and one during extension treatment). Among the 19 patients reporting suicidal ideation at baseline, 17 (90%) reported an improvement in suicidal ideation at last observation in the acute treatment period. Among the eight patients entering the extension phase who had reported suicidal ideation at baseline, all eight reported an improvement in suicidal ideation at last observation. As assessed by the CDRS-R item-13, emergence of suicidal ideation (worsening from baseline) occurred in two patients (2.8%) during the acute treatment phase and occurred in no patients during the extension treatment phase. One nonfatal suicide attempt occurred (SAE of self-injurious behavior/choking and suicidal ideation) and the patient was discontinued due to lack of efficacy. No completed suicides were reported during the study.
During the acute treatment phase, mean increases from baseline to end point in sitting systolic and diastolic blood pressure were 1.49 mmHg and 4.5 mmHg, respectively, while there was a mean decrease in heart rate (0.24 bpm) and a mean increase in weight (0.14 kg). For patients who entered the extension treatment phase, mean increases from baseline (last nonmissing screening visit) to end point in sitting systolic and diastolic blood pressure were 4.62 mmHg and 5.24 mmHg, respectively, while there was a mean decrease in heart rate (1.36 bpm) and a mean increase in weight (0.86 kg).
During the combined acute treatment and extension treatment periods, a total of 36/72 (50%) patients experienced PCS high blood pressure for at least one measurement with a total of 25/72 (35%) patients experiencing PCS high diastolic blood pressure and 26/72 (36%) patients experiencing PCS high systolic blood pressure. The range of treatment-emergent PCS systolic values was 115–142 (mean 128 mm Hg; median 127 mm Hg), and the range for treatment-emergent PCS diastolic values was 76 to 96 (mean 83 mm Hg; median 83 mm Hg). Among the patients experiencing treatment-emergent PCS high diastolic blood pressure, the baseline body mass index (BMI) range was 13–36, 72% (18/25) had baseline diastolic blood pressure above the 70th percentile, and 12% (3/25) had baseline diastolic blood pressure at or above the 95th percentile. Among the patients experiencing treatment-emergent PCS high systolic blood pressure, the baseline BMI range was 13–38, 92% (24/26) had baseline systolic blood pressure above the 70th percentile, and 42% (11/26) had baseline systolic blood pressure at or above the 95th percentile.
For most patients experiencing PCS high systolic or diastolic blood pressure, (21/36; 58%), the treatment-emergent PCS high blood pressure was transient (occurring and resolving during the study), while 15/36 patients (42%) experienced PCS high blood pressure at study end point. Among the 15 patients who experienced PCS high blood pressure at end point (10 diastolic and 9 systolic), the range of baseline blood pressures was from the 77th–95th percentile for diastolic blood pressure (70–78 mm Hg, one patient had baseline at the 95th percentile, baseline BMI range 13–36) and from the 52nd–97th percentile for systolic blood pressure (100–132 mm Hg, four patients had baseline at or above 95th percentile, baseline BMI range 13–31).
A total of 10 patients experienced sustained elevation (for three consecutive study visits) of PCS blood pressure; seven patients (five of whom were female) experienced sustained PCS elevation of diastolic blood pressure with a maximum value of 88 mm Hg, and five patients (three of whom were female) experienced sustained PCS elevation of systolic blood pressure with a maximum value of 142 mm Hg. Of the seven patients who experienced sustained PCS elevation in diastolic blood pressure, six had a PCS value at end point and had baseline blood pressures in the range of 80th–95th percentile (69–78 mm Hg, baseline BMI range 13–32), Of the five patients who experienced sustained PCS systolic blood pressure, all five had a PCS value at end point and had baseline blood pressures in the range of 52nd–95th percentile (100–120 mm Hg, baseline BMI range 13–29).
No patients reported fainting, syncope, or a blackout spell. No patients met electrocardiogram PCS criteria (interval >450 ms) for the corrected QT interval (QTc) using Fridericia's correction.
One patient experienced a PCS increase in hepatic enzymes (alanine transaminase/serum glutamic pyruvic transaminase >5 times upper limit of normal with total bilirubin within normal range) at the 14-week time point which returned to normal after 3 days while the patient remained on a stable duloxetine dose (30 mg). The PCS changes experienced by more than 5% of patients were observed for the following analytes during the acute treatment phase: Low hematocrit (13.3%), high creatine phosphokinase (8.7%), and high inorganic phosphorus (25.8%). During the extension treatment phase, the only laboratory value with a PCS change experienced by more than 5% of patients was high inorganic phosphorus (7.7%).
Among the four patients identified as CYP2D6 PMs, there were no reports of SAEs, discontinuations due to adverse events, PCS laboratory findings, or PCS electrocardiogram readings; however, one of the four PM patients met criteria for sustained elevation in systolic blood pressure during the study but did not meet criteria at end point.
Pharmacokinetics
All duloxetine plasma samples were obtained at steady-state, which was achieved after ∼3 days. As observed in adults, (Lobo et al. 2009) duloxetine plasma concentration-time data in pediatric patients were adequately described by a one-compartment pharmacokinetic model. The interpatient variability in duloxetine's oral clearance (CL/F) or oral volume of distribution (V/F) and the residual error were ∼50% for pediatric patients. Model evaluation using parameter sensitivity analysis showed that all parameters were estimated with adequate precision.
Patient characteristics such as age, body weight, creatinine clearance, CYP2D6 status, and menarche status did not have a statistically significant effect (p>0.05) on any of the duloxetine pharmacokinetic parameters, and age and body weight did not appear to affect duloxetine oral clearance (CL/F). In addition, dose did not influence duloxetine parameters; therefore, duloxetine pharmacokinetics were linear over the dose range of 20 to 120 mg. Gender did have a statistically significant influence (p<0.001) on CL/F. The CL/F of duloxetine in female patients was ∼30% lower than in male patients. Thus, on average, female patients were predicted to have ∼45% higher steady-state duloxetine plasma concentrations than males on the same dose. The median dose-normalized duloxetine concentration was similar in CYP2D6 EMs (0.55 ng/mL/mg, n=55) and CYP2D6 ultra metabolizers (0.48 ng/mL/mg, n=2), while the median dose-normalized duloxetine concentrations were nearly 3-fold higher in CYP2D6 PMs (1.69 ng/mL/mg, n=4). However, there was considerable overlap in the observed concentrations for CYP2D6 PMs and EMs.
Duloxetine CL/F was ∼42%–60% higher in pediatric patients than in adult patients, and the volume of distribution was 16% lower in pediatric patients than in adult patients; as a result, the estimated half-life of duloxetine in pediatric patients was ∼50% shorter than in adult patients (∼12 hours, Cymbalta Full Prescribing Information 2011).
Comparison of model-predicted duloxetine concentrations at steady state showed that the median steady-state duloxetine concentration-time profile was lower in the pediatric population relative to adults. The average steady-state duloxetine concentration was 37% lower in the pediatric population (30 mg: Median 16.7 ng/mL; 60 mg: 33.5 ng/mL) than in adults (30 mg: Median 26.5 ng/mL; 60 mg: 52.9 ng/mL). The range of duloxetine plasma concentrations in the pediatric population was encompassed within the range of plasma concentrations observed in adults.
Effectiveness
Secondary assessments included in this study were a global severity measure (CGI-Severity) and a symptom severity measure (CDRS-R). The mean changes (SD) in the CGI-Severity score from baseline to end point were −2.11 (1.17) and −2.7 (1.07) for acute and extension treatment phases, respectively, and median changes were −2.0 for both the acute and the extension treatment phases. The mean changes in CDRS-R scores from baseline to end point were −35.4 (SE: 1.0) (MMRM) and −35.8 (SD: 10.3) (OC) for the acute treatment phase and −39.4 (SE: 0.5) (MMRM) and −40.1 (SD: 9.2) (OC) for the extension treatment phase. A total of 43/72 (59.7%) patients achieved remission at end point during the 18-week acute treatment phase.
Discussion
The primary purpose of this study was a preliminary assessment of the safety, tolerability, and pharmacokinetics of duloxetine in children and adolescents with MDD. A total of 72 patients was enrolled in this open-label study, 81% completed the 10-week dose titration phase, 67% completed 18 weeks of acute treatment, and 58% of patients completed 30-weeks of treatment. Completion rates in this study were comparable to other pediatric MDD studies.
The flexible dosing scheme employed in this study allowed for clinical and pharmacokinetic assessments over a wide range of duloxetine doses from 20 to 120 mg once daily. Escalation from initial doses of 20 mg or 30 mg once daily was to follow good clinical practice such that the dose should have been escalated only if tolerated and only if response was not optimal at the current dose. Results of this study showed that most patients (76%) required dose escalation to higher duloxetine doses (≥60 mg once daily) to optimize effectiveness. Consistent with the observation that most study patients required higher doses of duloxetine, the primary population pharmacokinetic findings from this study showed that the duloxetine clearance in children and adolescents was nearly twice that observed in adults; consequently, duloxetine half-life in children and adolescents was approximately half that observed for adults.
In the current pediatric study, gender was the only covariate that had a statistically significant effect on duloxetine clearance; however, associations between clinical safety measures by gender and duloxetine pharmacokinetics could not be systematically evaluated based on this small, uncontrolled study. Of note, sustained elevation in blood pressure and TEAEs and SAEs were more frequently observed in females than males, though discontinuation due to an adverse event occurred with the same frequency in females and males. In contrast, more males (16/37; 43.2%) experienced transient PCS changes in laboratory values compared with females (8/35; 22.9%).
Regarding safety, the most frequently reported TEAEs in this study were similar to those observed in duloxetine-treated adult patients with MDD; however, the incidence of individual TEAEs was lower than in adult MDD studies of duloxetine. (Hudson et al. 2005) The lower incidence of TEAEs may, in part, be related to the lower starting dose (20 or 30 mg once daily) and slower titration to an effective dose (10-week titration phase) than employed in most adult MDD studies of duloxetine (60 mg once daily initial and fixed dose, or 1-week titration to doses >60 mg once daily). While higher mean changes in blood pressure were observed in this study (e.g., diastolic blood pressure increase of ∼5 mm Hg) compared with results from adult MDD studies (1–2 mm Hg) (Hudson et al. 2005), it should be noted that this study did not include a placebo or active control. Many patients (50%) experienced treatment-emergent PCS elevation in systolic or diastolic blood pressure; and, for many of the patients who experienced PCS elevation in blood pressure (58%), the PCS elevation was transient and resolved during the study. However, 15 patients experienced PCS elevation in blood pressure at end point. Among patients who experienced treatment-emergent PCS elevation in blood pressure at end point, the majority (>90%), had baseline blood pressures above the 70th percentile based on age, height, and weight; therefore study-defined PCS criteria would be met if patients had at least an increase of 5–9 mm Hg, and such increases in blood pressure (>5 mm Hg) may be clinically significant for some patients. Given the variability in pediatric blood pressure measurements, placebo-controlled studies will be needed to better characterize the effect of duloxetine on blood pressure in pediatric patients. However, as part of good clinical practice, it is reasonable to monitor blood pressure when treating pediatric patients with drugs that may increase noradrenergic neurotransmission.
Changes in suicidal ideation and behavior were closely monitored during the study using the C-SSRS. Of patients with suicidal ideation reported at baseline, ∼90% had improvement in suicidal ideation by end point. As measured by the C-SSRS, two patients (2.8%) experienced worsening of suicidal ideation or behavior during the study, which is relatively consistent with other large meta-analyses reporting an incidence of spontaneously reported suicidality in the range of 0% to 8.3% in antidepressant treatment arms and 0% to 5.4% in placebo treatment arms in pediatric antidepressant studies. (Hammad et al. 2006)
The results of this study must be interpreted with regard to several limitations. This was an uncontrolled, open-label study. Further larger-scale, well-controlled studies are needed to better characterize the safety, efficacy, and pharmacokinetics of duloxetine in the treatment of children and adolescents with MDD. In particular, the effect on clinical outcomes of the shorter observed duloxetine half-life in pediatric patients than adults and the differential clearance by gender could not be systematically evaluated based on the results of this uncontrolled study. Additionally, patients with significant medical or psychiatric co-morbid conditions were excluded from this study, and the study patient characteristics reflected minimal co-morbid medical and psychiatric conditions and restricted concomitant medication use. Therefore, the safety results from this study have limited generalizability to a broader population with greater medical and psychiatric burdens.
Conclusion
It has been suggested that a failure to conduct and use information from appropriately designed preliminary pharmacokinetic and dose-finding studies has, in part, contributed to the inability of several antidepressants, with known efficacy in adults, to show efficacy in large-scale, phase 3, clinical trials in the pediatric MDD population. (Atuah et al. 2004; Findling et al. 2006) The current study was designed to address the aforementioned concern.
Clinical Significance
Results from the current study suggest that pediatric patients generally tolerated and experienced improvement of MDD symptoms with duloxetine 30 to 120 mg once daily treatment, although many patients did experience transient PCS elevations in blood pressure. Because body weight and age did not have a statistically significant effect on duloxetine pharmacokinetics, the study results suggest that differential dosing based on body weight or age is not warranted within the pediatric patient population. Results also support that differential dosing is not warranted for pediatric patients relative to adults. The results of this preliminary study have informed the design of larger, well-controlled studies, currently underway, to better understand the benefits and risks of duloxetine in the treatment of children and adolescents with MDD.
Footnotes
Disclosures
Ms. Prakash, Ms. Pangallo, Ms. Quinlan, and Drs. Lobo, Tamura, and Bullok are employees and stockholders of Eli Lilly and Company. Dr Kratochvil was supported by NIMH Grant 5K23MH06612701A1; received grant support from Eli Lilly, Abbott, Somerset, Shire; was a consultant for Eli Lilly, Abbott, Neuroscience Education Institute, MedAvante, AstraZeneca, Theravance, Seaside, Quintiles, and Pfizer; is editor of the Brown University Child & Adolescent Psychopharmacology Update; is a member of the REACH Institute Primary Pediatric Psychopharmacology Steering Committee; receives study drug from Eli Lilly and Abbott for an NIMH-funded study. Dr. Emslie received research support Biobehavioral Diagnostics Inc., Eli Lilly, Forest Laboratories, GlaxoSmithKline, and Somerset; has been a consultant for Biobehavioral Diagnostics Inc., Eli Lilly, Forest Laboratories, GlaxoSmithKline, INC Research Inc., Lundbeck, Pfizer Inc., Seaside Therapeutics Inc, Shire, Validus Pharmaceuticals, and Wyeth Pharmaceuticals; and has been on the Speakers Bureau for Forest Laboratories. Dr. March has served as a consultant or scientific advisor to Pfizer, Lilly, GlaxoSmithKline, BMS, Johnson and Johnson, Psymetrix, Attention Therapeutics, Avanir, Alkermes, Translational Venture Partners, LLC, Vivus, and MedAvante; received study drug for an NIMH-funded study from Eli Lilly and from Pfizer; is an equity holder in MedAvante; receives royalties from Guilford Press, Oxford University Press, and MultiHealth Systems. Dr. March receives research support from Pfizer, NIMH, NIDA, SRI, and NARSAD. Dr. March has not engaged in promotional work, e.g., speakers bureau or training, for over 15 years. Dr. March's conflict of interest is fully reported to the University, viewable at
Acknowledgments
The authors would like to thank the patients and their caregivers for participating in this trial; Jeff Dinkel and Christina Lux for their contributions to implementing this trial; Mary Pat Knadler for bioanalytical support; Sarah Kogut and Melissa Spann for statistical analysis review; Angela Lorio for editorial support.