
Abstract
Objective:
To evaluate safety and tolerability of four doses of immediate-release molindone hydrochloride in children with attention-deficit/hyperactivity disorder (ADHD) and serious conduct problems.
Methods:
This open-label, parallel-group, dose-ranging, multicenter trial randomized children, aged 6–12 years, with ADHD and persistent, serious conduct problems to receive oral molindone thrice daily for 9–12 weeks in four treatment groups: Group 1—10 mg (5 mg if weight <30 kg), group 2—20 mg (10 mg if <30 kg), group 3—30 mg (15 mg if <30 kg), and group 4—40 mg (20 mg if <30 kg). The primary outcome measure was to evaluate safety and tolerability of molindone in children with ADHD and serious conduct problems. Secondary outcome measures included change in Nisonger Child Behavior Rating Form-Typical Intelligence Quotient (NCBRF-TIQ) Conduct Problem subscale scores, change in Clinical Global Impressions-Severity (CGI-S) and -Improvement (CGI-I) subscale scores from baseline to end point, and Swanson, Nolan, and Pelham rating scale-revised (SNAP-IV) ADHD-related subscale scores.
Results:
The study randomized 78 children; 55 completed the study. Treatment with molindone was generally well tolerated, with no clinically meaningful changes in laboratory or physical examination findings. The most common treatment-related adverse events (AEs) included somnolence (n=9), weight increase (n=8), akathisia (n=4), sedation (n=4), and abdominal pain (n=4). Mean weight increased by 0.54 kg, and mean body mass index by 0.24 kg/m2. The incidence of AEs and treatment-related AEs increased with increasing dose. NCBRF-TIQ subscale scores improved in all four treatment groups, with 34%, 34%, 32%, and 55% decreases from baseline in groups 1, 2, 3, and 4, respectively. CGI-S and SNAP-IV scores improved over time in all treatment groups, and CGI-I scores improved to the greatest degree in group 4.
Conclusions:
Molindone at doses of 5–20 mg/day (children weighing <30 kg) and 20–40 mg (≥30 kg) was well tolerated, and preliminary efficacy results suggest that molindone produces dose-related behavioral improvements over 9–12 weeks. Additional double-blind, placebo-controlled trials are needed to further investigate molindone in this pediatric population.
Clinical trials.gov Identifier:
NCT00626236.
Introduction
Molindone, a first-generation, typical antipsychotic agent, was recently studied in a pediatric population with schizophrenia (Treatment of Early-Onset Schizophrenia Spectrum Disorders) (Sikich et al. 2008). At a mean dose of 60 mg/day, given with benztropine (1.0 mg/day), this agent had a favorable side-effect profile compared with risperidone and olanzapine and comparable efficacy. Greenhill and colleagues (1985) conducted a double-blind, parallel-group study of molindone compared with thioridazine in a group of antipsychotic-naïve children aged 6–11, who were hospitalized for undersocialized conduct disorder, aggressive type. Molindone, at a mean dose of 27 mg/day, provided significant improvements in the 10-item Conners' Rating Scale, the Inpatient Aggression Scale, and the hostility, antisocial, and violence subscale measures from the Children's Psychiatric Rating Scale in comparison with the first placebo period and was equally efficacious to thioridazine. No significant differences in treatment-emergent side effects were noted between the two treatments (Greenhill et al. 1985).
The present proof-of-concept study evaluated the safety and tolerability of four dosages of a capsular immediate-release formulation of molindone 5–40 mg/day given in three divided doses in children with ADHD and persistent, serious conduct problems. The decision to use an open-label study design without a placebo control was due to the primary goal of assessing safety and tolerability in our first study evaluating molindone in this vulnerable population of 6- to 12-year-old children.
Methods
This phase 2a, open-label, parallel-group, dose-ranging study was conducted at 10 sites in the United States from October 2008 to September 2009. All study procedures complied with the Declaration of Helsinki and U.S. Code of Federal Regulations dealing with clinical studies. Each subject's parent or legally authorized representative provided written informed consent, and each subject provided written assent. The protocol was approved by the institutional review board at each participating site.
Subjects
The study population was male or female children, aged 6–12 years, who had a diagnosis of ADHD with persistent serious conduct problems evidenced by a baseline score of ≥27 on the Disruptive Behavior Disorder (DBD) subscale of the Nisonger Child Behavior Rating Form–Typical Intelligence Quotient (NCBRF-TIQ) Version, and a score of ≥2 on at least one of the following three items of the Conduct Problem subscale of the NCBRF-TIQ: (7) knowingly destroys property, (22) gets in physical fights, or (30) physically attacks people. The diagnosis of ADHD was confirmed by using the Schedule for Affective Disorders and Schizophrenia for School-aged Children—Present and Lifetime Versions (Kaufman et al. 1997).
Eligible subjects were required to weigh at least 16 kg, have an IQ ≥71, be free of antipsychotics for at least 2 weeks before the baseline visit, and be receiving a stable dose of a U.S. Food and Drug Administration (FDA)-approved psychostimulant (methylphenidate or amphetamine) for at least 30 days before baseline. Subjects were excluded for a current or lifetime diagnosis of bipolar disorder, posttraumatic stress disorder, personality disorder, or psychotic disorder or if currently meeting diagnostic criteria for major depressive disorder, obsessive compulsive disorder, or pervasive developmental disorder, or any other anxiety disorder as a primary disorder, as defined in the Diagnostic and Statistical Manual of Mental Disorders, 4th edition, text revision (DSM-IV-TR) (American Psychiatric Association 2000). Subjects were otherwise in good physical health.
Study schedule
The study consisted of a screening period (28 days before the first dose), a titration period (2–5 weeks), a maintenance period (6 weeks), and a safety follow-up (30 days after the final study visit) (Fig. 1). Subjects were randomized to one of four treatment groups at visit 1 of the titration period (baseline visit). Each subject participated in the study for 16–19 weeks, depending on treatment group assignment, and the study took place over 11 months.
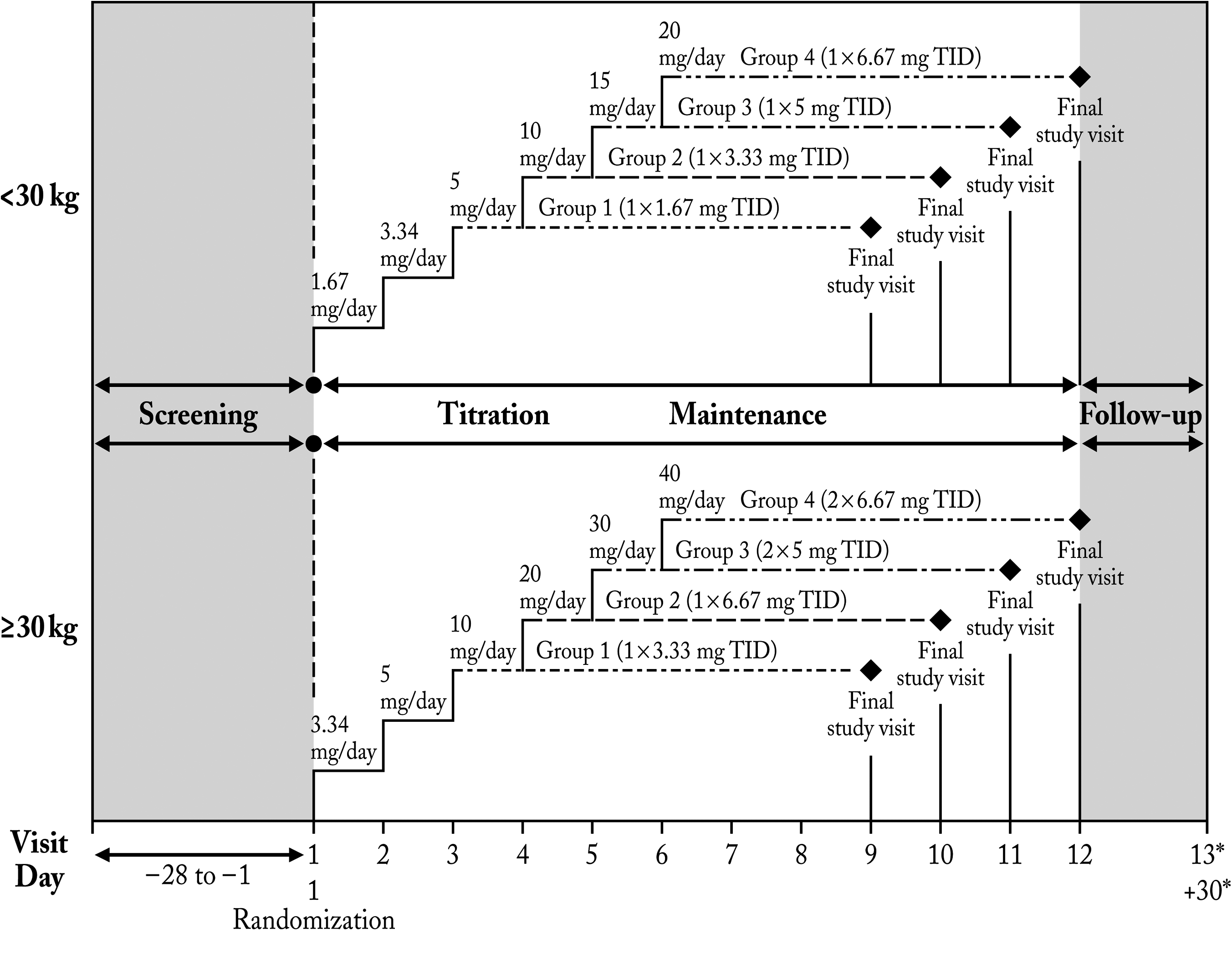
Study design. *Safety follow-up phone visit—30 days post final study visit.
During the screening period, subjects underwent a physical examination, electrocardiography, and the Kaufman Brief Intelligence Test, Second Edition (Naugle et al. 1993), if an IQ test was not performed within the 12 months before screening.
At visit 1 (baseline visit) during the titration period, subjects were randomized according to weight into one of four treatment groups, each with two dose levels of molindone hydrochloride (Moban® 2010; Endo Pharmaceuticals Inc., Chadds Ford, PA) (Fig. 1). Subjects weighing <30 kg were given 1.67 mg/day for 1 week and 3.34 mg/day for 1 week followed by 5 mg/day maintenance dosing (group 1) or continued weekly titration up to 10 mg/day (group 2), 15 mg/day (group 3), or 20 mg/day (group 4). Subjects weighing ≥30 kg were given 3.34 mg/day dose for 1 week and 5 mg/day for 1 week followed by 10 mg/day maintenance dosing (group 1) or continued weekly titration up to 20 mg/day (group 2), 30 mg/day (group 3), or 40 mg/day (group 4). The titration schedule was followed until the target dose was reached. Daily doses were given in three administrations ∼8 hours apart because of the short elimination half life of the drug (∼2 hours) (2010).
During the maintenance period, subjects remained at the target dose for 6 weeks. Subjects in the highest-dose group only (group 4) were permitted to down-titrate once at the investigator's discretion in consultation with the medical monitor. A poststudy follow-up consisted of a phone call to each subject 30 days after the final study visit.
After randomization, subjects returned to the study site weekly for 8–11 visits (2–5 weeks of titration and 6 weeks of maintenance) for dose increases, adverse event (AE) monitoring, physical evaluations such as vital signs and weight measurements, and the administration of safety and efficacy scales. In addition, blood and urine for clinical laboratory tests were collected at screening, maintenance week 2, and final visit, and concomitant medications were assessed.
Treatment adherence was not formally evaluated, but records of study medication, doses administered, and intervals between visits were maintained and monitored during this study. Investigators were to use their discretion to assess nonadherence and take appropriate action as necessary (e.g., discontinuation from study).
Outcome measures
The primary outcome measure was safety and tolerability of molindone. Secondary outcome measures included change in the NCBRF-TIQ Conduct Problem subscale score (Aman et al. 2008) from baseline to end point and change in scores in Clinical Global Impressions–Severity (CGI-S) and –Improvement (CGI-I) subscales (Guy 1976) from baseline to end point and the Swanson, Nolan, and Pelham rating scale–revised (SNAP-IV) (Swanson et al. 2001) ADHD-related subscale scores.
Safety
Safety was evaluated at every visit and included assessments of treatment emergent AEs and neurological side effects as assessed by changes in the Simpson-Angus scale (Simpson and Angus 1970), Barnes Akathisia scale (Barnes 1989), and Abnormal Involuntary Movement Scale (AIMS) (Guy 1976). Safety was also assessed through clinical laboratory tests, vital signs, physical examinations, and electrocardiography.
NCBRF-TIQ
A newly validated version of the NCBRF, the NCBRF-TIQ excludes those items from the NCBRF questionnaire that are specific to children with developmental disabilities (Aman et al. 2008). The NCBRF-TIQ was completed by the same parent or legally appointed representative when possible at each visit. The questionnaire characterizes any special circumstances or mediating factors that may have affected the child's recent behavior and scores the child's social behaviors at home and problem behaviors. NCBRF-TIQ ratings are grouped into a single Positive Social subscale and the 6 Problem Behavior subscales: Overly Sensitive, Oppositional Behavior, Conduct Problem, Hyperactivity, Inattention, and Withdrawal/Dysphoria. The Oppositional Behavior and Conduct Problem subscale totals are combined to form the DBD-Total score, and the Hyperactivity and Inattention subscales are combined to form the ADHD-Total score.
CGI scale
This scale represents the clinician's view of a subject's global functioning before and after administration of a study medication (Guy 1976). Severity of Illness (CGI-S) and Global Improvement (CGI-I) are both CGI subscales rated 1 to 7, with 7 being “extremely ill” or “very much worse,” respectively. Therapeutic success is indicated by a lower overall score in subsequent testing.
SNAP-IV scale
The SNAP-IV rating scale assesses 18 symptoms of ADHD and 8 symptoms of oppositional defiant disorder (ODD) as specified in the DSM-IV-TR and the International Statistical Classification of Diseases and Related Health Problems, 10th Revision (Swanson et al. 2001). A severity estimate is assigned to each symptom, from 0 (not at all) to 3 (very much). The SNAP-IV rating scale was completed by the same parent or legally appointed representative when possible at each visit. SNAP-IV ratings are grouped into 3 subscales: ADHD-Inattention, ADHD-Hyperactivity/Impulsivity, and ODD. In addition, the first two subscales are combined to form the ADHD-Combined subscale. The ADHD-Inattention, ADHD-Hyperactivity/Impulsivity, and ADHD-Combined subscale scores were secondary efficacy variables.
Data analysis
Notably, efficacy assessment was not a primary objective in this study, and a target sample size of 72 subjects was not based on any statistical considerations, but was judged adequate to provide safety and efficacy information for a proof-of-concept study. Therefore, efficacy evaluations were carried out by using numeric trends suggestive of a response to treatment, and tests of statistical significance were performed as an adjunct to this assessment and to assist in the planning of future studies.
Changes from baseline in NCBRF-TIQ, CGI-S, and SNAP-IV scores, as well as actual CGI-I scores, at each postbaseline visit and end point were compared between treatment groups by using the analysis of variance test (for the overall comparisons). Statistical t-test was used for pairwise comparison in the intent-to-treat population, defined as all randomized subjects who received at least one dose of study medication with at least one postbaseline efficacy assessment. Subjects who had discontinued the study without postrandomization scores were not included in the analysis of efficacy variables. The last-observation-carried-forward technique was used in the efficacy analysis for subjects who discontinued the study with postrandomization scores.
Results
Subject demographics and disposition
Subject disposition is presented by treatment group in Figure 2. There were 94 subjects screened for entry into the study. Of these, 16 subjects were screening failures, and 78 were randomized into the study. Of the 78 subjects randomized, 55 (70.5%) completed the study, and 23 (29.5%) discontinued. In this analysis, the safety and intent-to-treat (ITT) populations were the same. Table 1 presents demographic and baseline characteristics of study subjects, and Table 2 presents concomitant or previous medical conditions. The majority of subject were male (n=67, 85.9%) and white (n=43, 55.1%); African Americans comprised 42.3% of the population (n=33). The mean age of study subjects was 8.8 years and the mean weight was 33.58 kg. Treatment groups did not vary in baseline characteristics except for a higher percentage of females, greater mean weight, and somewhat longer durations of ADHD or ODD symptomatology, as assessed by the patients' past medical history, in group 3.
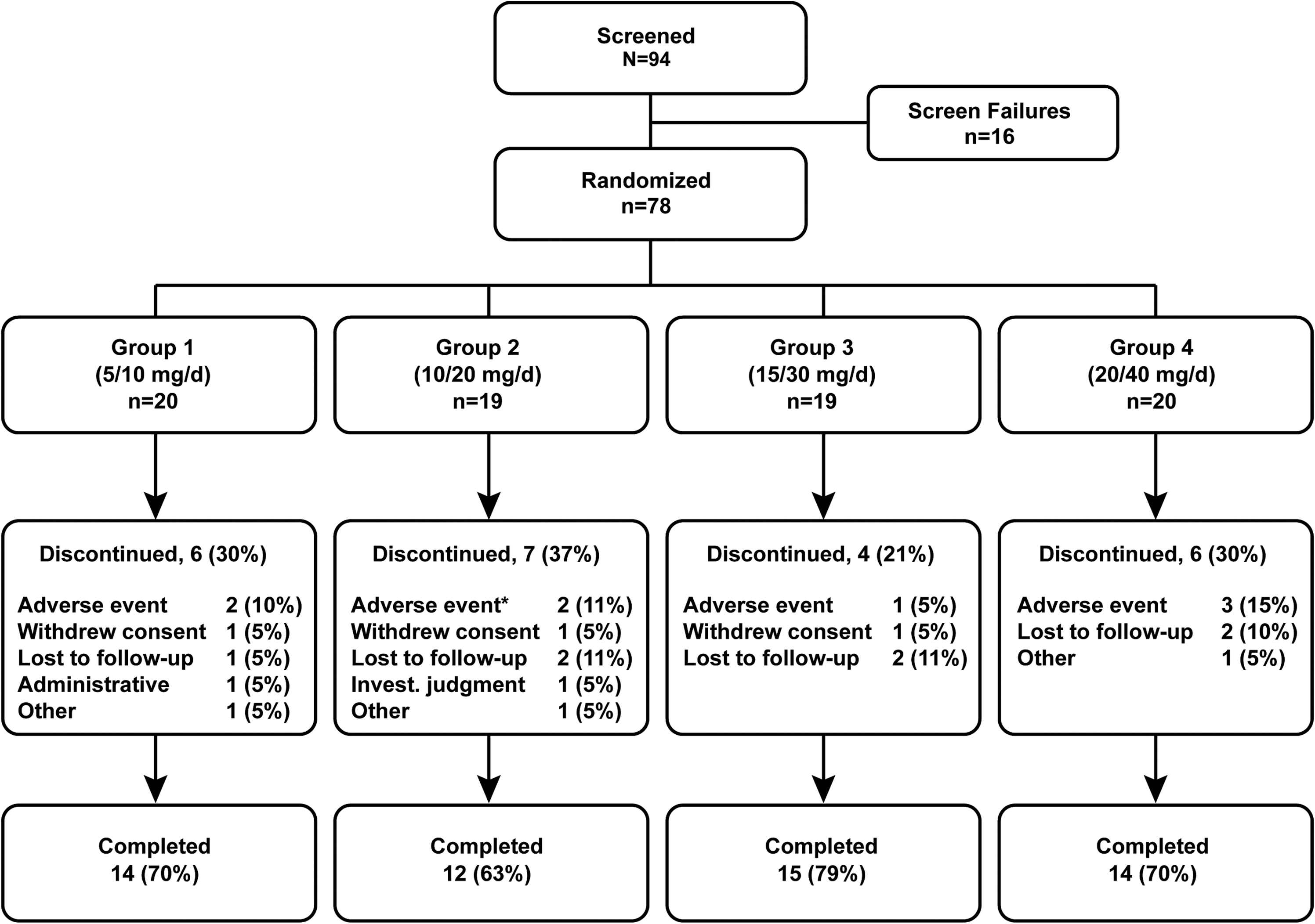
Patient disposition, n (%). *One additional patient in group 2 withdrew due to an adverse event; however, the subject's primary reason for discontinuation was withdrawn consent/assent.
Min, max=minimum, maximum; SD=standard deviation.
ADHD=attention-deficit/hyperactivity disorder; ODD=oppositional defiant disorder; SD=standard deveation; N/A=not applicable.
Subject enrollment per site ranged from 2 to 16, with a mean enrollment of 7 subjects per site.
Subjects were concurrently treated with psychostimulants ranging from total daily doses of Adderall®/Adderall XR® 5–40 mg, Concerta® 18–54 mg, Focalin XR® 10–20 mg, Vyvanse® 20–70 mg, methylphenidate 15–20 mg, Metadate CD® 20–40 mg, and Daytrana® 20–30 mg. Subjects were prohibited from taking concomitant antipsychotics during the study and were also not to receive antidepressants, hypnotics, anticonvulsants, antihypertensives, or antihistamines, unless prescribed to treat a treatment emergent AE. Benztropine, a cholinergic antagonist, was permitted for the treatment of emergent extrapyramidal symptoms (EPS), and was administered to one subject to treat akathisia.
Safety
Adverse events
There were 61 subjects (78.2%) with one or more AEs (Table 3), the vast majority of which were mild to moderate in severity. The most common AEs by preferred term were somnolence (n=10, 12.8%), abdominal pain (n=9, 11.5%), and weight increase (n=8, 10.3%). AEs were least common in groups 1 (n=12, 60.0%) and 2 (n=15, 78.9%) and most common in groups 3 (n=17, 89.5%) and 4 (n=17, 85.0%).
AE=adverse event.
There were 45 subjects (57.7%) with one or more AEs considered either possibly or probably related to study medication according to the investigator's opinion. The most common treatment-related AEs included somnolence (n=9, 11.5%), weight increase (n=8, 10.3%), akathisia (n=4, 5.1%), sedation (n=4, 5.1%), and upper abdominal pain (n=4, 5.1%). As with the overall incidence of AEs, the incidence of related AEs was lowest in group 1, intermediate in group 2, and most common in the higher-dose groups.
Nine subjects discontinued due to AEs (Fig. 2) including one discontinuation in group 2 that was reported as the subject's withdrawal of consent. There was no apparent association between discontinuation of study medication and the dose administered. In these nine subjects, the AEs leading to discontinuation were dizziness (n=2), abdominal pain/pain (n=2), akathisia/psychomotor hyperactivity (n=2), and agitation/abnormal behavior (n=3).
Of the 20 subjects in group 4, 15 continued treatment as planned, and 5 underwent a dosage reduction during the course of the study. AEs that led to these dose reductions included somnolence (n=2), akathisia (n=1), dyspepsia (n=1), and sedation (n=1). One patient had a treatment interruption due to muscle spasm.
There were no deaths. Two subjects had serious AEs; however, neither was considered to be related to study medication.
Laboratory results and physical exam findings
No clinically meaningful changes occurred in standard hematology or clinical chemistry values. Increases in prolactin were observed in a small number of subjects. The overall mean prolactin level was 5.2 μg/L at baseline and 7.0 μg/L at visit 7. Patients in groups 3 and 4 had greater mean increases in prolactin levels between baseline and visit 12 (3.2 and 3.7 μg/L, respectively) compared with subjects in groups 1 and 2 (1.6 and −1.6 μg/L, respectively) during that same interval. Although eight subjects had shifts in prolactin levels from “normal” to “high” at visit 7, by visit 12, only 3 subjects (two in group 3 and one in group 4) continued to have “normal” to “high” shift levels of prolactin. No clinically significant changes were noted in vital signs, electrocardiography results, or physical exam findings.
Modest increases occurred in weight and body mass index (BMI) over the course of treatment. Between baseline and the final visit, the overall mean change in weight was 0.54 kg, and the overall mean increase in BMI was 0.24 kg/m2. Both the changes in weight and BMI appeared to increase with dose (Table 4). Eight subjects (10.3%) reported weight gain as an AE; by convention, this was considered to be a weight increase of >7% during treatment.
BMI=body mass index; bpm=beats per minute.
Extrapyramidal symptoms
Extrapyramidal symptoms (EPS) were evaluated by using three scales (the AIMS, Barnes Akathisia, and Simpson-Angus scales). At any visit during the study, not more than five subjects overall (or more than three in any treatment group) had an individual subscore, on any of the three scales, that worsened compared with baseline. Nine subjects had EPS reported as AEs. Akathisia and psychomotor hyperactivity, which were felt to be related to study medication, resulted in discontinuation of one patient each, respectively, from the trial.
Efficacy: NCBRF-TIQ
Figure 3 shows mean changes from baseline in the NCBRF-TIQ Conduct Problem subscale score, the primary efficacy outcome measure for the study. The mean score for the NCBRF-TIQ Conduct Problem subscale at baseline was 20.4 in group 1, 25.7 in group 2, and 26.0 in both groups 3 and 4. Scores decreased from baseline in all treatment groups (Table 5), thus indicating improvement. By visit 12 (final study visit), the mean changes from baseline were −7.0, −8.7, −8.2, and −14.3 in groups 1, 2, 3, and 4, respectively. Notable decreases (improvement) in mean scores occurred as early as visit 3 in groups 2, 3, and 4; whereas improvements in group 1 occurred later, at visit 6 and thereafter.
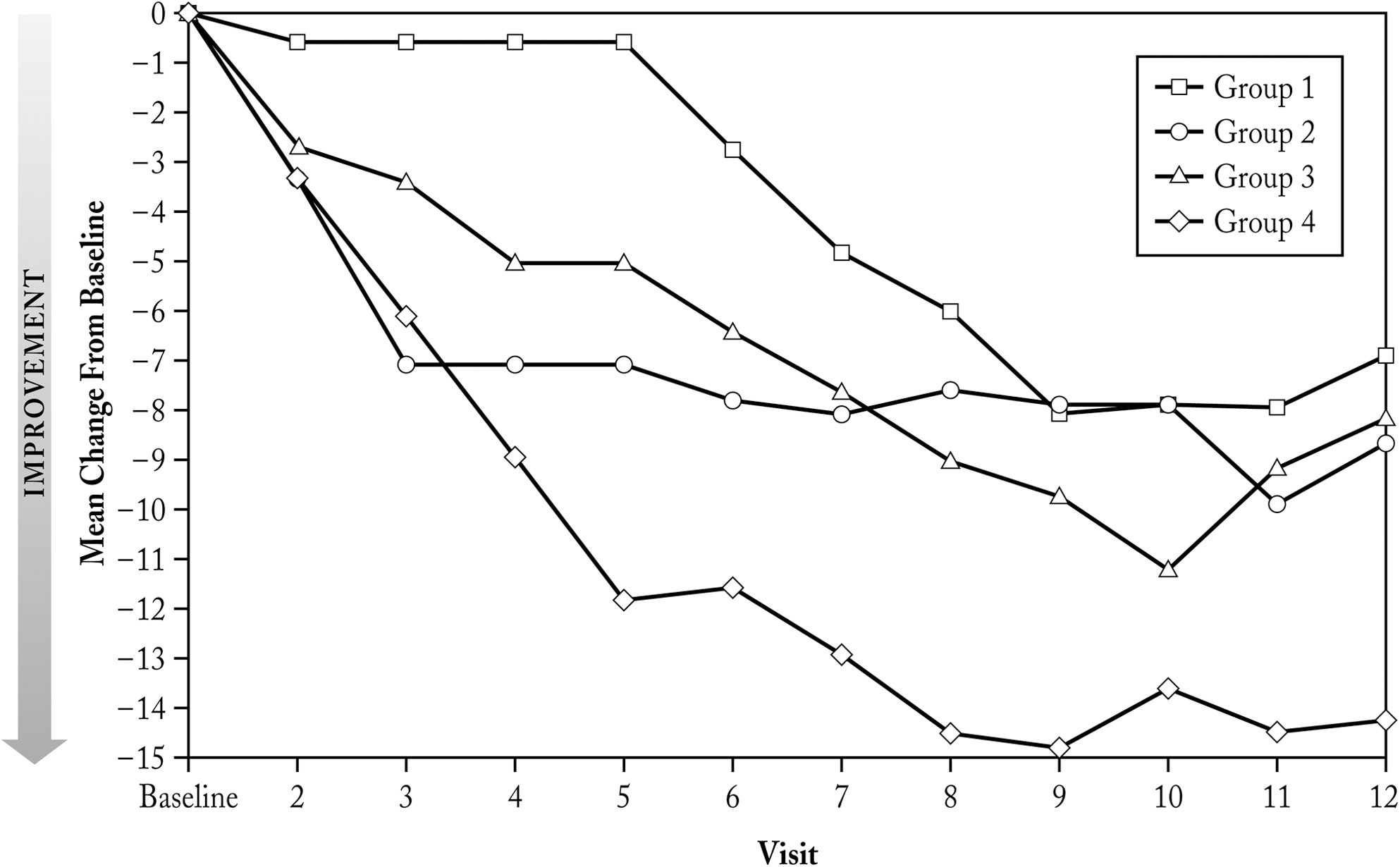
NCBRF-TIQ Conduct Problem Subscale–Mean Change from Baseline (ITT Population). NCBRF-TIQ=Nisonger Child Behavior Rating Form-Typical Intelligence Quotient; ITT=intent-to-treat.
NCBRF-TIQ=Nisonger Child Behavior Rating Form-Typical Intelligence Quotient.
Efficacy: Secondary outcomes
CGI scale
At baseline, CGI-S scores were 4.35, 4.73, 4.63, and 4.70 in groups 1, 2, 3, and 4, respectively. These scores decreased (improved) over time in all treatment groups. At visit 12 (final study visit), mean changes from baseline were −1.00, −1.00, −1.26, and −1.70 in groups 1, 2, 3, and 4, respectively. Improvements in CGI-S scores were seen in group 4 earlier than in the other treatment groups. Similarly, improvements in CGI-I scores were also seen earlier in group 4 compared with the other groups.
SNAP-IV scale
SNAP-IV subscale scores improved, as evidenced by decreased scores in all groups after visit 2 compared with baseline. Mean changes in the ADHD-Inattention subscale score were −4.35, −6.26, −6.84, and −8.15 in groups 1, 2, 3, and 4 at week 12, respectively. The respective changes in the ADHD-Hyperactivity/Impulsivity subscale scores at week 12 were −5.55, −5.89, −5.42, and −8.50. Likewise, the ODD Subscale scores improved with molindone treatment in groups 1, 2, 3, and 4 with reductions of −5.55, −6.47, −5.21, and −9.35, respectively. Across these SNAP-IV subscales, the greatest improvements were consistently found in the highest dose group (group 4).
Discussion
Treatment with molindone in this study targeted serious conduct problems and aggressive symptoms (i.e., physical fights, physical attacks on others, and intentional destruction of property) that were present in children with ADHD despite ongoing stabilized treatment with a psychostimulant. Although these aggressive symptoms are not considered a part of the core symptoms of ADHD, they may well be a part of the clinical picture when constituent symptoms or the full syndrome of a comorbid DBD are present. Although the current study did not require a DBD diagnosis in the pediatric patients with ADHD included here (only 44% had a DBD diagnosis), primarily because of the exploratory nature of this proof-of-concept study, the required baseline score of ≥27 on the DBD subscale of the NCBRF-TIQ and a score of ≥2 on at least one of three items of the Conduct Problem subscale were thought to be able to accurately identify patients with impulsive aggression and/or serious conduct problems. Our investigation of these constructs in patients with ADHD are supported by a recent consensus report on impulsive aggression by Jensen and colleagues (2007), who stated that “given the current state of knowledge, for the purposes of pursuing pharmacological indications, impulsive aggression should be studied principally within well-defined patient groups, with well-established psychiatric disorders, such as ADHD, PTSD, autism, and/or bipolar disorder” (Jensen et al. 2007). Although impulsive aggression may be considered nonspecific in the current U.S. diagnostic nosology, it is considered serious enough to warrant a specific therapeutic intervention (Pappadopulos et al. 2006).
A recent meta-analysis of 28 studies demonstrated that successful stimulant treatment can be expected to reduce overt and covert aggression in children with ADHD (Connor et al. 2002), and similar results have been reported more recently (Blader et al. 2010). Given that the patients in our study had an average baseline ADHD score of ∼40, as measured on the SNAP-IV-Combined subscale, it could be argued that psychostimulant treatment may not have been fully optimized before randomization. Still, widely variable effect sizes characterize stimulant efficacy on aggression, and beg the question of what to do with the considerable number of children whose severe aggressive behaviors do not respond to ADHD treatment. When you also consider that having fewer symptoms of ADHD has been found to be a predictor of improvement in boys with conduct disorder (Lahey et al. 2002), we acknowledge that future studies should be designed with this consideration.
ADHD is recognized as a chronic disorder. The presence of persistent serious comorbid conduct problems in these patients generally signals a more severe form of ADHD. In a 10-year longitudinal study, serious conduct problems persisted for about 36% of young adults in whom ADHD had been diagnosed and who had exhibited significant conduct problems as youths. These young adults had significantly increased risks for psychoactive substance use disorders, smoking, and bipolar disorders, as well as higher rates of expulsions from school, crime convictions, sexual intercourse before the age of 16, and being fired from a job (Biederman et al. 2008). Therefore, an early intervention that is effective in treating these comorbid conduct problems could minimize the deleterious impact of the disorder and its attendant chronic social dysfunction within this group of children (Wilens et al. 2002).
The poor outcomes (e.g., depression, antisocial or criminal behavior) associated with the diagnosis of severe forms of aggression are sufficiently common and serious to warrant the evaluation of a medication with the adverse effect profile of molindone. Since there are no FDA-approved treatments for serious persistent conduct problems or impulsive aggression for children, this potential therapy can satisfy a critical unmet medical need. Although very preliminary, the results from this investigation encourage the further study of molindone as a suitable pharmacological tool to address refractory aggression in optimally stimulant-treated children and adolescents with ADHD.
In adults with schizophrenia, it has been noted that second-generation antipsychotics have a reduced propensity, when compared with first-generation antipsychotics, to cause tardive dyskinesia (Correll et al. 2004; Kane and Correll 2010). As noted in a review by Correll et al. (2004), however, few of the existing studies were designed to focus on tardive dyskinesia and its accurate identification. A recent paper concludes that the incidence of tardive dyskinesia appears relatively unchanged since the 1980s (Woods et al. 2010). Unfortunately, it is unknown whether there is a difference between these two classes of drugs with regard to the development of tardive dyskinesia in children and teenagers due to the paucity of comparative data. This is an important gap in knowledge when one considers that patients included in this study are commonly seen in clinical practice, and the risk of tardive dyskinesia with long-term molindone treatment (or other first-generation antipsychotics) in this patient population has not yet been characterized. Although molindone is generally considered a first-generation, or typical, antipsychotic, its gene expression profile in a model of retinal cell lines is markedly different than that of other typical antipsychotics (Polymeropoulos et al. 2009).
The main limitations of the current open-label study are the absence of masking and a placebo-controlled arm. The open-label nature of this study allowed both clinicians and subjects (and their caregivers) to be aware of the treatment assigned, which may contribute to bias. Another limitation is the brevity of the study in view of the chronicity that may be associated with impulsive aggression. Therefore, all the efficacy findings should be considered preliminary, although the time course of the effect is not suggestive of a typical placebo response (Mayberg et al. 2002). The short duration of this study does not allow for an assessment of longer-term safety, and given the small sample size, the power to detect adverse effects of molindone is limited.
Conclusions
Molindone showed clinical benefit with an acceptable side-effect profile in this study. The drop out rate of 11.5% due to AEs was lower than that observed in the double-blind study conducted by Sikich and colleagues (2008) comparing molindone, risperidone, and olanzapine in childhood schizophrenia and schizoaffective disorder. In that study, 20% of molindone-treated patients discontinued due to AEs. The wider molindone dose range used (10–140 mg/day) may have contributed to this finding. Although the primary objective of this phase 2a open-label study was not to assess efficacy of molindone, the robustness and the consistency of the efficacy supports existing evidence that pharmacotherapy may be indicated in the treatment of children with serious persistent conduct problems or problematic, impulsive aggression (Findling 2003). Considering the gravity of the condition and the putative benefits seen in this short-term, open-label study, further investigation of the use of molindone in the treatment of this population appears appropriate, including its efficacy and safety over the long term.
Clinical Significance
Children with ADHD and persistent conduct problems face potential serious consequences and require careful treatment. This study demonstrated that treatment with molindone hydrochloride (at daily doses of 10–40 mg/day for patients weighing ≥30 kg and 5–20 mg/day in patients weighing <30 kg) was safe and generally well tolerated. This study shows that acute treatment with molindone hydrochloride may result in dose-related behavioral improvements.
Disclosures
Dr. Findling receives or has received research support, acted as a consultant and/or served on a speaker's bureau for Abbott, Addrenex, AstraZeneca, Biovail, Bristol-Myers Squibb, Forest, GlaxoSmithKline, Johnson & Johnson, KemPharm, Lilly, Lundbeck, Merck, Neuropharm, Novartis, Noven, Organon, Otsuka, Pfizer, Rhodes, sanofi-aventis, Schering-Plough, Seaside Therapeutics, Sepracor, Shire, Solvay, Sunovion, Supernus Pharmaceuticals, Transcept, Validus, and Wyeth. Ms. Stocks, Dr. Taneja, and Dr. Baroldi are employees of Supernus Pharmaceuticals, Inc. Supernus Pharmaceuticals, Inc. provided financial and material support, monitoring, data collection and management, and data analysis to the authors and study investigators. Jennifer Stocks prepared the first draft of the manuscript with critical input and revisions by all the authors for important intellectual content.
Footnotes
Acknowledgments
The authors thank John Simmons, M.D., and the staff of Peloton Advantage, LLC, for their editorial assistance with the preparation of the final draft.