
Abstract
Objective:
Information on psychostimulant treatment in long-term studies for attention-deficit/hyperactivity disorder (ADHD) in adolescents is limited. This study aimed to assess the safety and effectiveness of lisdexamfetamine dimesylate (LDX) over 52 weeks in adolescents with ADHD.
Methods:
This open-label multicenter study enrolled eligible participants after their participation in a randomized, double-blind, placebo-controlled 4 week trial in adolescents with ADHD. Following a 4 week dose-optimization phase, participants were maintained on treatment for up to ∼48 weeks on an optimal dose. Safety assessments included treatment-emergent adverse events (TEAEs), vital signs, laboratory findings, and electrocardiograms. Effectiveness measures included the ADHD Rating Scale IV (ADHD-RS-IV; primary) and Clinical Global Impressions-Improvement (CGI-I). The Youth Quality of Life-Research Version (YQOL-R) was also included in this study; raw scores are transformed to a 0–100 point scale.
Results:
Of 269 enrolled (from the antecedent study), 265 (98.5%) were in the safety population and effectiveness population. Common TEAEs (≥5%) with LDX included upper respiratory tract infection (21.9%), decreased appetite (21.1%), headache (20.8%), decreased weight (16.2%), irritability (12.5%), insomnia (12.1%), nasopharyngitis (7.2%), influenza (6.8%), dizziness (5.3%), and dry mouth (5.3%). At end point, for all LDX doses in the overall safety population, mean (SD) increase from baseline in systolic blood pressure was 2.3 (10.53) mm Hg, diastolic blood pressure was 2.5 (8.37) mm Hg, and pulse rate was 6.3 (12.74) bpm. No clinically meaningful electrocardiogram or vital sign changes were observed. At end point with LDX treatment, the ADHD-RS-IV mean (SD) total score change from antecedent study baseline was −26.2 (9.75) (p<0.001); 87.2% of participants were improved (CGI-I=1 or 2). Baseline (antecedent study) mean (SD) YQOL-R perceptual total score was 79.8 (11.28) and increased by 3.9 (9.73) at end point (p<0.001).
Conclusions:
LDX demonstrated a long-term safety profile similar to that of other long-acting psychostimulants and was effective, as indicated by improvements in ADHD symptoms and participant-perceived YQOL, in adolescents with ADHD.
Clinical Trial Registration:
NCT00764868,
Introduction
Based on the percentage of adolescents who met the Diagnostic and Statistical Manual of Mental Disorders, 4th ed., Text Revision (DSM-IV-TR) (American Psychiatric Association 2000) ADHD criteria, there are ∼3,500,000 in the United States with ADHD (Schubiner and Katragadda 2008). Adolescents may exhibit less hyperactivity than do children with ADHD. However, when compared with their peers, adolescents with ADHD have increased impulsivity (e.g., “making irresponsible choices”), increased inattention, and a high rate of psychiatric comorbidities (Schubiner and Katragadda 2008). When compared with treated adolescents, untreated adolescents with ADHD had higher rates of academic failure, emotional problems, earlier onset of substance abuse, delinquency, teenage pregnancy, sexually transmitted infections, and poor driving records (Schubiner and Katragadda 2008).
There are several efficacious treatment options for ADHD. Psychostimulant medications are the most commonly prescribed and are recommended as first-choice ADHD treatment options by various guidelines (Pliszka and AACAP Work Group on Quality Issues 2007; Wolraich et al. 2007; Graham and Coghill 2008). However, there are only a few long-term studies (Hoare et al. 2005; Hammerness et al. 2009; Findling et al. 2010) to date that have assessed the safety and effectiveness of psychostimulant use in adolescents. Hence, there is a need for long-term studies assessing psychostimulant use, especially long-acting formulations.
Lisdexamfetamine dimesylate (LDX), a long-acting prodrug stimulant, is indicated by the United States Food and Drug Administration for ADHD treatment of children (6–12 years), adolescents (13–17 years), and adults. Two long-term, open-label LDX studies conducted in children (Findling et al. 2008) and adults (Weisler et al. 2009) with ADHD demonstrated a safety profile consistent with that of other psychostimulant medications. In the child study, mainly mild to moderate treatment-emergent adverse events (TEAEs) were experienced by 78% of participants (Findling et al. 2008). Common TEAEs with an incidence >5% with LDX treatment included decreased appetite (33%), headache (18%), decreased weight (18%), insomnia (17%), upper abdominal pain (11%), upper respiratory tract infection (11%), irritability (10%), nasopharyngitis (10%), vomiting (9%), cough (7%), and influenza (6%). In the adult study, with all LDX doses, TEAEs (mainly mild to moderate severity) were experienced by ∼88% of participants (Weisler et al. 2009). TEAEs with an incidence ≥5% with any LDX dose included upper respiratory tract infection (21.8%), insomnia (19.5%), headache (17.2%), dry mouth (16.6%), decreased appetite (14.3%), irritability (11.2%), anxiety (8.3%), nasopharyngitis (7.4%), sinusitis (6.6%), decreased weight (6.0%), back pain (5.4%), and muscle spasms (5.2%).
Additionally, LDX demonstrated efficacy in adolescents with ADHD and a safety profile consistent with that of long-acting psychostimulants in a randomized, double-blind, parallel-group, placebo-controlled, forced-dose titration study (4 weeks) (Findling et al. 2011). This is a first report of the long-term effects of LDX in adolescents (aged 13–17 years) with at least moderately symptomatic ADHD. Of note, this study included an examination of adolescents' perception of quality of life (QOL) during LDX treatment.
Methods
Study design and participants
This open-label extension study was conducted across 45 sites in the United States from November 2008 to April 2010. Adolescents 13–17 years of age (inclusive at the time of the antecedent study entry) with a primary diagnosis of ADHD who participated in an antecedent parallel-group, double-blind, placebo-controlled, fixed-dose titration, short-term (4 weeks) LDX study (Findling et al. 2011) and were treated with LDX or placebo, were eligible to participate in this long-term study, if they completed a minimum of 3 weeks of the antecedent study and were not terminated for noncompliance, adverse events (AEs), or other safety reasons. Participants remained blinded to prior treatment (placebo or LDX) assignments in the present study, although it was an open-label design. In order to preserve the blind of the antecedent study, investigators in the present study were blinded to data from the antecedent study; however, to facilitate patient management, a few sites requested unblinded data after all participants had completed participation in the antecedent study and after all treatment in the current study had been initiated.
Key study inclusion criteria for the antecedent study included being 13–17 years of age (inclusive) at the time of consent; a total score of ≥28 on the ADHD Rating Scale IV (ADHD-RS-IV) (DuPaul et al. 1998) at baseline; age-appropriate intellectual function; blood pressure (BP) measurements ≤95th percentile for age, sex, and height; and a negative pregnancy test for females. Also included were participants with no clinically relevant abnormalities based on their medical history or physical examination, and those and their parent(s) or legally authorized guardian(s) who were willing and able to adhere to protocol requirements.
Key exclusion criteria for the antecedent study included comorbid psychiatric disorder and/or symptoms/conditions that might contraindicate treatment with LDX and influence safety or effectiveness analyses; a concurrent chronic or acute illness or an unstable medical condition that might confound the safety results; clinically significant electrocardiogram (ECG) or history of serious cardiac problems (e.g., cardiovascular disease, advanced arteriosclerosis, structural cardiac abnormality, cardiomyopathy, serious heart rhythm abnormalities, coronary artery disease, and family history of sudden cardiac death or ventricular arrhythmia) prior to treatment; suicidal ideations; substance abuse; and/or allergy, hypersensitivity, and/or intolerance to amphetamine. Additionally, participants who were nonresponsive to amphetamine in the antecedent study or were receiving prohibited medications that have central nervous system effects or affect performance (e.g., sedating antihistamines, decongestant sympathomimetics) prior to the antecedent study were not eligible to participate. Participants were also not eligible to participate in this study if they were terminated from the antecedent 4 week study because of noncompliance and/or an AE for which continued treatment would be medically contraindicated.
This study was designed to assess the safety and effectiveness of a daily morning dose (30, 50, or 70 mg/day) of LDX in adolescents with ADHD. It consisted of a 4 week dose-optimization phase followed by a 48 week, open-label maintenance phase and a safety follow-up phase. Regardless of prior treatment in the antecedent study, all participants began this open-label study with a 30 mg/day dose of LDX. During the optimization phase, beginning at week 0 and extending through week 4, participants were titrated to an optimal dose of medication with site visits occurring every 7 days (±2 days). Titration was based on overall response, defined as acceptable, ineffective, or intolerable. An acceptable response to LDX treatment was defined as a ≥30% reduction in ADHD-RS-IV total score from baseline (visit 0 of antecedent study) and a Clinical Global Impressions-Improvement (CGI-I) rating of 1 or 2 with tolerable side effects, whereas an ineffective response was defined as not meeting these criteria. For an ineffective response, the LDX dose was titrated upward to the next dose strength. An intolerable response was defined as occurrence of side effects that were characterized as intolerable by the investigator, and the dose was decreased to a lower dose (if available). If at the lower dose, intolerable side effects persisted, the participant was removed from the study. To obtain the optimal dose of LDX, the dose could be decreased or increased by 20 mg/day increments (maximum dose of 70 mg/day), based on investigator judgment. During the maintenance phase (48 additional weeks), participants returned for study visits at 28 day intervals (±5 days). The optimized LDX dose was continued, or, if deemed necessary by the investigator, further dose adjustments were possible. A safety follow-up telephone call occurred 7 days (±2 days) after the last LDX dose. Baseline demographics were obtained during the antecedent trial. Baseline safety and effectiveness assessments used for the current study were those obtained at the baseline visit of the antecedent study.
Voluntarily signed informed consent was provided by a parent or legally authorized guardian, and participants signed documentation of assent to indicate that they were aware of the study procedures and restrictions. This study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice E6 according to the International Conference on Harmonisation (ICH) guidelines. In addition, the institutional review board of each institution reviewed and approved the informed consent form and the protocol.
Safety measures
Safety measures included reported AEs, physical examinations, height and weight assessments, vital signs, a 12 lead ECG, and clinical laboratory tests. AEs were coded using the Medical Dictionary for Regulatory Activities Version 11.1 (MedDRA 2009). Ongoing AEs from the antecedent study were recorded as medical history. All study AEs were recorded from the time of signed informed consent to the follow-up call, were assessed for relatedness and severity by the investigator at all study visits, and were summarized by system organ class and preferred term for each LDX dose group. If an AE increased in intensity relative to the antecedent study during this open-label study, it was considered a new TEAE. TEAEs were defined as AEs that started or worsened at any time from the first day of LDX treatment in this study to the third day (inclusive) after treatment had stopped. Whether an AE was treatment related or considered serious was determined by the investigator. Serious AEs (SAEs) were defined as clinical occurrences that were judged by the investigator to be “medically important” and that resulted in death, were life threatening, required or prolonged hospitalization, resulted in persistent or significant disability, or resulted in a congenital defect. A severe AE was defined as an AE that was incapacitating, resulting in an inability to work or complete usual activity.
Physical examinations were performed at week 4/end point of the antecedent study, at week 52, and end point/early termination (ET) of this study. Height was measured at week 4/end point of the antecedent study and, in the current study, at week 4 of the dose-optimization phase and at weeks 12, 20, 28, 36, 44, and 52/or ET end point of the maintenance phase. Weight was measured and vital signs (including sitting systolic BP [SBP], diastolic BP [DBP], and pulse) obtained at all study visits. A 12 lead ECG was recorded at week 4 and end point/ET of the antecedent study and at weeks 12, 24, 36, and 52 and at end point/ET of the maintenance phase. Clinical laboratory tests were assessed at week 4/end point of the antecedent study and at week 20 and week 52 and at end point/ET in this study. The baseline values for height and weight, vital signs, laboratory tests, and ECG were the baseline of the antecedent study, and visit 1 values for the current study were those from week 4/end point of the antecedent study. Clinical significance of ECG and laboratory assessment results was determined by the investigator. Medication adherence was assessed from week 1 to week 52 and at end point/ET of the current study. Reasons for discontinuation from the study were AEs, protocol nonadherence/participant noncompliance, refusal to participate further, loss to follow-up, lack of effectiveness, and other.
Effectiveness measures
The primary measure of effectiveness was the clinician-reported ADHD-RS-IV (DuPaul 1998). The key effectiveness end point of the study was the change from baseline (defined as baseline from the 4 week study) of the ADHD-RS-IV total score at end point. ADHD-RS-IV was also assessed at each postbaseline visit. The scale consists of 18 items that evaluate ADHD symptom levels based on current DSM-IV-TR criteria and is divided into two subscales (inattention and hyperactivity/impulsivity) of nine items each. Items are scored on a four point scale that ranges from 0 (never, rarely) to 3 (very often). The range of possible scores is 0–54, with higher scores indicating more impairment. The ADHD-RS-IV was completed by the same rater at each visit, whenever possible, and information was obtained from the participant's parent/legal guardian.
The clinician-reported CGI evaluated global illness severity (CGI-S) and improvement (CGI-I) over time (Guy 1976). The CGI-S assessed global illness severity based on a seven point scale, which ranges from 1 (normal, not at all ill) to 7 (among the most extremely ill). The CGI-I, which evaluated global improvement from week 1 of the dose-optimization phase up to 48 weeks or end point/ET of the maintenance phase, is also based on a seven point rating scale, ranging from 1 (very much improved) to 7 (very much worse). Improvement was assessed relative to baseline of the antecedent study. Clinician-reported scales (i.e., ADHD-RS-IV and CGI), were completed when possible by the same rater (clinician) experienced in evaluating adolescents with ADHD.
The QOL of adolescents participating in the study was assessed using the Youth Quality of Life Instrument-Research Version (YQOL-R) (YQOL-R 2002; Edwards et al. 2002; Patrick et al. 2002). The participant-reported YQOL-R is a validated, 56 item instrument consisting of two modules: contextual items (potentially verifiable by others) and perceptual items (known only to the participant). The perceptual module is reported herein and is categorized into four general domains (self, relationship, environment, and general QOL). A total of five scores can be generated from the perceptual items, a total perceptual score and four individual domain scores. The YQOL-R raw score is transformed to a 0–100 point scale to assist in result interpretation, with higher scores indicating better QOL. The YQOL-R was completed by the participants at study entry, at week 28, and at week 52 and end point/ET of the current study.
Safety analyses
Safety was reported for all enrolled participants who had taken at least one dose of LDX treatment. The change from baseline in safety parameters was from baseline of the antecedent study. End point was defined as the last on-treatment visit after the first visit in the current study. The last valid assessment after baseline was used for participants with early termination.
Vital signs, ECG results, physical examination findings, and laboratory evaluations were summarized using descriptive statistics. Outlier criteria were defined a priori. At end point/ET, numbers of participants who met defined outlier criteria were recorded. Outlier criteria for SBP included SBP ≥120 mm Hg and, separately, SBP ≥120 mm Hg that constituted an increase from baseline of ≥10 mm Hg. Outlier criteria for DBP included DBP≥80 mm Hg and, separately, DBP ≥80 mm Hg that constituted an increase from baseline of ≥10 mm Hg. Outlier criteria for pulse rate included pulse ≥100 bpm and, separately, pulse ≥100 bpm that constituted an increase from baseline of ≥15 bpm.
Mean z scores for height and weight were determined based on Centers for Disease Control and Prevention (CDC) (Centers for Disease Control and Prevention 2000) reference values for these measurements. Participants who took 80–100% of the LDX treatment from visit to visit were considered clinically adherent to treatment. Treatment compliance was assessed based on capsule counts. The adherence rate was calculated as (number of capsules dispensed−number of capsules returned)/(number of capsules prescribed per day×number of days in treatment period) ×100. Statistically, a participant was considered to be protocol compliant if between 80% and 120% (inclusive) compliance with LDX treatment was maintained throughout the study.
The clinician-administered Columbia-Suicide Severity Rating Scale (C-SSRS) (Posner et al. 2009) was included after this study was begun (week 24/next scheduled visit to week 52 and end point/ET), and safety monitoring based on review of responses was used to assess suicidal ideations and behaviors.
Effectiveness analyses
The effectiveness population (full analysis set [FAS]) was defined as all enrolled participants who took at least one dose of LDX treatment and had one postenrollment assessment of the primary effectiveness measure (ADHD-RS-IV total score). Changes from baseline (from antecedent 4 week study) in ADHD-RS-IV total score at each visit and end point/ET were summarized and assessed using a two-sided paired t test. The ADHD-RS-IV total and subscale scores were summarized by visit and dose. The secondary effectiveness outcome measures also included CGI-I and the YQOL-R.
For analysis of CGI-I, scores were dichotomized into those globally improved (CGI-I ratings of 1 [very much improved] or 2 [much improved]) and those not improved (CGI-I ratings of 3–7). Summary statistics were presented at each postbaseline visit of this study and at end point/ET. For the YQOL-R, only perceptual items were included in the a priori statistical plan for analysis; paired t tests were used to evaluate the change from baseline (4 week antecedent double-blind study) to week 28, week 52, and end point/ET.
Results
Participant disposition and demographics
Of the 314 participants who were enrolled and randomized in the antecedent double-blind 4 week study, 310 participants received at least one dose of LDX or placebo treatment and were included in that study's safety population. Of the 310 participants, 257 (82.9%) completed the antecedent double-blind study. There were 269 participants from the antecedent study who enrolled in the current study, of which 265 (98.5%) were included in the safety and FAS populations. Of the 269 participants, 198 (73.6%) received LDX treatment and 71 (26.4%) received placebo in the antecedent study; randomization in the antecedent study was 1:1:1:1 for the LDX 30 mg/day, 50 mg/day, and 70 mg/day dose groups and placebo, respectively (Findling et al. 2011). A total of 156 (58.0%) participants completed the current study
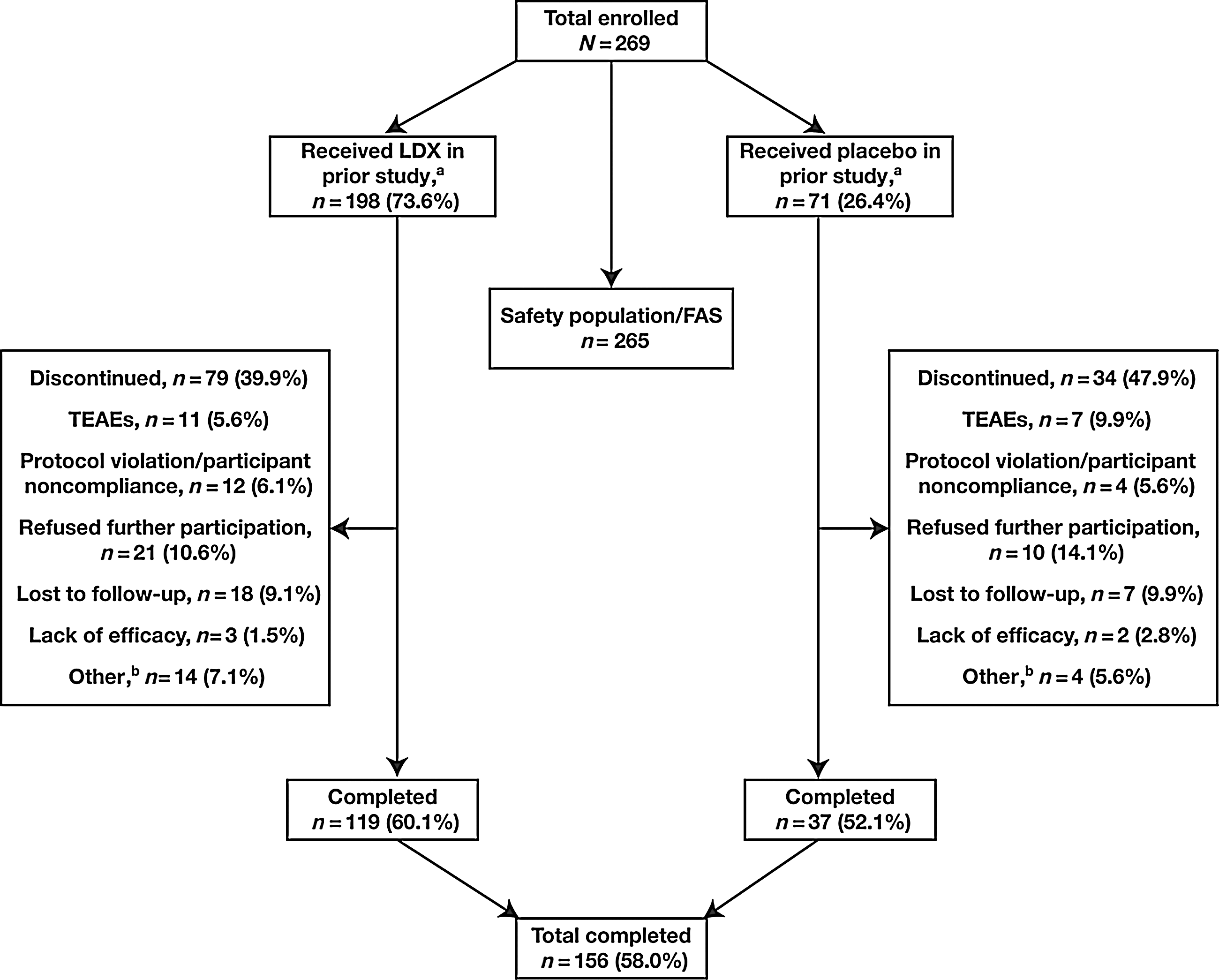
Participant disposition. aParticipants received treatment in the antecedent double-blind study. bA total of 14 participants were discontinued in the prior LDX-treated group for other reasons: Pregnancy (n=3), moved or were out of area (n=6; one was sponsor requested), took prohibited medication for an AE (n=1), withdrew consent (n=1), had a misuse/compliance issue with study drug (n=1), met exclusion criteria (n=1), and tested positive on urine drug screen (n=1). Four participants were discontinued in the prior placebo-treated group for other reasons: Moved or were out of area (n=3) and tested positive on urine drug screen (n=1). FAS, full analysis set; LDX, lisdexamfetamine dimesylate; TEAEs, treatment-emergent adverse events.
Demographic characteristics for the safety population are summarized in Table 1. Overall, most participants were male (70.6%), white (79.6%), and non-Hispanic or non-Latino (86.4%). At baseline, the majority of participants were moderately or markedly ill (95.1%), based on CGI-S ratings. Moreover, most participants were diagnosed with inattention (34.7%) or combined (64.5%) ADHD subtypes, and the mean (SD) time since initial ADHD diagnosis was 4.6 (4.14) years prior to the screening visit of the 4 week double-blind study. No apparent differences existed in baseline and demographic characteristics between the LDX- and placebo-treated groups.
Demographics and baseline characteristics were carried forward from the antecedent 4 week double-blind study.
ADHD-RS-IV, Attention-Deficit/Hyperactivity Disorder Rating Scale IV; CGI-S, Clinical Global Impressions-Severity; LDX, lisdexamfetamine dimesylate; QOL, quality of life; SD, standard deviation; YQOL-R, Youth Quality of Life Instrument-Research Version.
Treatment dosing
There were 55 participants who had a final LDX dose of 30 mg/day, 101 participants who had a final dose of 50 mg/day, and 109 who had a final dose of 70 mg/day. During the study, of 150 participants who were titrated to a maximum dose of 70 mg/day, 109 participants had a final dose of 70 mg/day, whereas 34 and 7 participants had been down-titrated to the 50 mg/day and 30 mg/day dose levels, respectively. Of the 87 participants titrated to a maximum LDX dose of 50 mg/day, 67 were on that same dose at final visit, and 20 were down-titrated to the 30 mg/day dose. At week 52 (n=158), the percentage of participants in the 30 mg/day group was 20.9%, in the 50 mg/day group it was 38.6%, and in the 70 mg/day group it was 40.5%.
Safety
Of the overall safety population, 230 (86.8%) participants experienced TEAEs; for participants who had previously received LDX and placebo, 169 of 195 (86.7%) and 61 of 70 (87.1%), respectively, reported TEAEs. Common TEAEs reported by ≥5% of participants are summarized in Table 2. The incidence of TEAEs ≥5% by actual LDX dose suggests a dose-dependent increase in the percentage of participants with TEAEs. Most TEAEs were mild to moderate in severity. There were 13 (4.9%) participants who experienced 18 severe TEAEs (i.e., incapacitating AEs). These severe TEAEs included dizziness (2), headache (2), migraine (2), aggression (1), agitation (1), dermatitis contact (1), ectopic pregnancy (1), hydrocele (1), joint sprain (1), pelvic fracture (1), pneumonia (1), testicular torsion (1), traumatic liver injury (1), weight decrease (1), and wrist fracture (1). There were no deaths during the study. Moreover, there were 10 (3.8%) participants who reported 15 serious TEAEs (i.e., clinical occurrences judged by the investigator to be “medically important”). These serious TEAEs included syncope (4), aggression (2), ectopic pregnancy (1), hydrocele (1), joint sprain (1), pelvic fracture (1), pneumonia (1), testicular torsion (1), traumatic liver injury (1), vasovagal syncope (1), and wrist fracture (1). No serious TEAEs were considered related to treatment except for three syncopal episodes. In this trial, syncope was considered an important medical event requiring reporting as an SAE; there were 5 such events (4 syncope, 1 vasovagal syncope) in four participants. Of these 5 events, 2 (both syncope) were mild and 3 (2 syncope, 1 vasovagal syncope) were moderate, and all resolved without any intervention or sequelae. Of the three moderate events, the one episode of moderate vasovagal syncope occurred in a participant treated with LDX who had a prior history of a structural cardiac abnormality and mitral insufficiency by echocardiogram. On sponsor-determined early termination (because of a protocol violation), the patient was not eligible for study participation and she subsequently reported an SAE of syncope. Vasovagal syncope occurred while taking 70 mg/day of LDX on day 21 of treatment in the current study (participant had received 70 mg/day of LDX in the antecedent study) and resolved without treatment. Per protocol, this was considered a treatment-related and serious AE. TEAEs resulted in early termination in 15 of 265 (5.7%) participants. These 15 participants had 19 TEAEs that led to early termination, including depressed mood (3), insomnia (3), aggression (2), abdominal pain (2), depression (1), ectopic pregnancy (1), increased BP (1), irritability (1), paranoia (1), suicidal ideation (1), tic (1), visual hallucination (1), and weight decrease (1).
LDX, lisdexamfetamine dimesylate; MedDRA, Medical Dictionary for Regulatory Activities; TEAEs, treatment-emergent adverse events.
SBP, DBP, and pulse rate for the overall safety population are summarized in Table 3. There were increases from baseline (of the antecedent study) in SBP, DBP, and pulse rate at weeks 12, 24, 36, and 52 and at end point (Table 3). At end point, for all LDX doses in the overall safety population, mean (SD) increase in SBP was 2.3 (10.53) mm Hg, DBP was 2.5 (8.37) mm Hg, and pulse rate was 6.3 (12.74) bpm from baseline (Table 3). For each LDX dose group, moderate increases were observed in mean (SD) change from baseline in SBP, DBP, and pulse rate with time. There was no clear relationship between the various doses and the mean change; however, this study was not statistically designed or powered to assess such relationships among doses. The mean (SD) increases in SBP (mm Hg) at end point by actual LDX doses (30, 50, and 70 mg/day) were similar: 3.1 (9.15), 2.1 (11.49), and 2.0 (10.31), respectively. For DBP (mm Hg), the mean (SD) changes from baseline at end point were comparable: 2.0 (7.73), 3.1 (7.70), and 2.2 (9.27) for the 30, 50, and 70 mg/day actual LDX doses, respectively. When the LDX doses were considered in the same order, the increases in mean (SD) pulse rate (bpm) at end point were 5.7 (11.92), 4.2 (12.21), and 8.5 (13.37).
Baseline is from the antecedent 4 week study.
ET, early termination; SD, standard deviation.
In the safety population, 33 (12.5%) participants met SBP outlier criteria of ≥120 mm Hg with an increase from baseline of ≥10 mm Hg, and 4 (1.5%) had an SBP measure of ≥140 mm Hg at study end point. For DBP, 20 (7.5%) participants met outlier criteria of ≥80 mm Hg with an increase of ≥10 mm Hg, and there were no participants with a value of ≥90 mm Hg at study end point/ET. There were 11 (4.2%) participants who met pulse outlier criteria at end point/ET of ≥100 bpm with an increase from baseline of ≥15 bpm, and none had a pulse rate of ≥120 bpm on two consecutive visits.
There were no ECG findings at end point that were deemed clinically significant by the investigator. At study end point (n=257), the mean (SD) change from baseline (n=265; antecedent study) in heart rate was 5.2 (12.24) bpm, with a final value of 75.3 (12.63). A total of 12 (4.7%) of 257 participants met heart rate outlier criteria of ≥100 bpm. Based on the ICH clinical evaluation guidelines suggesting that QTcF interval may be considered a useful and relevant correction for assessing participants who receive stimulant treatment and subsequently have increases in heart rate, the QTcF interval was used for ECG analysis (United States Department of Health and Human Services 2005). At end point, the mean (SD) change from baseline for the QTcF interval was 1.8 (17.19) ms. During an unscheduled posttermination visit (7 days posttermination), one participant had a QTcF of 464 ms and an increase from baseline of 71 ms, meeting QTcF outlier criteria of ≥450 ms and change from baseline of ≥60 ms. This participant had an AE of elevated QTcB interval (453 ms) prior to initiating treatment in the current study. The AE was considered unrelated to treatment, was unresolved, and led to early termination from the study 6 days after commencing treatment with LDX 30 mg/day. This participant had received placebo in the antecedent study.
Height and weight were converted to mean (SD) z scores based on the CDC growth charts data by age and sex (Centers for Disease Control and Prevention 2010). For weight at baseline, the mean (SD) z score was 0.6 (0.88) and, at end point, was 0.3 (0.86). Also, by absolute weight, from baseline to end point, the mean (SD) changes in weight (kg) by dose were −0.1 (3.91), −0.4 (4.80), and −1.9 (6.08) for the 30, 50, and 70 mg/day dose groups, respectively. At baseline, the mean (SD) z score for height was 0.2 (0.95) and, at end point, was 0.2 (0.94). Overall mean (SD) change in height (cm) from baseline at end point was 2.5 (2.55). Mean (SD) z score for body mass index (BMI) was 0.6 (0.82) at baseline and was 0.2 (0.86) at end point. Changes in z scores for weight and BMI of study participants were numerically small at study end point, and actual values remained slightly above the mean for the age- and sex-matched general population. The observed weight and BMI decreases were consistent with known effects of psychostimulants. Analysis of shift in BMI indicated that of the 26 participants classified as obese at baseline, 10 participants remained classified as obese at end point. Based on CDC-published growth chart criteria for classification of weight status, of the 171 participants classified as healthy weight at baseline, 5 were underweight at end point/ET. There were no underweight participants at baseline.
Other safety assessments included physical examinations, clinical laboratory evaluation (biochemistry, hematology, and urinalysis), and the C-SSRS. There were no clinically notable physical examination findings. Moreover, the majority of participants at both screening of the antecedent study and end point of the current study had normal physical examination findings. Overall, changes in mean laboratory measures were generally modest, and there were no clinically notable trends over time. The C-SSRS was administered to 52 participants who were still actively enrolled at week 24. No new suicidal ideations or behaviors were reported by study participants during this study based on semistructured interview items that captured the occurrence, severity, and frequency of suicide-related thoughts and behaviors.
Effectiveness
Mean (SD) ADHD-RS-IV (primary effectiveness measure) total score at baseline from the antecedent 4 week study was 38.0 (7.00) (Fig. 2). The change from baseline in mean (SD) ADHD-RS-IV total score at end point was −26.2 (9.75), indicating significant improvement in ADHD symptoms with LDX treatment (p<0.001) (Fig. 2). Significant decreases in ADHD-RS-IV total scores were achieved at all postbaseline weeks with LDX treatment (p<0.001) (Fig. 2). There were no significant between-group differences in mean changes from baseline to end point in ADHD-RS-IV total scores based on original group assignments from the antecedent study. For the ADHD-RS-IV inattention and hyperactivity/impulsivity subscales, the mean (SD) baseline scores were 22.6 (3.35) and 15.4 (6.19), respectively. The mean (SD) changes in the inattention and hyperactivity/impulsivity subscale scores at end point, from baseline of the antecedent 4 week study, were −15.1 (6.05) and −11.1 (5.89), respectively, and were significant at all postbaseline visits (p<0.001).
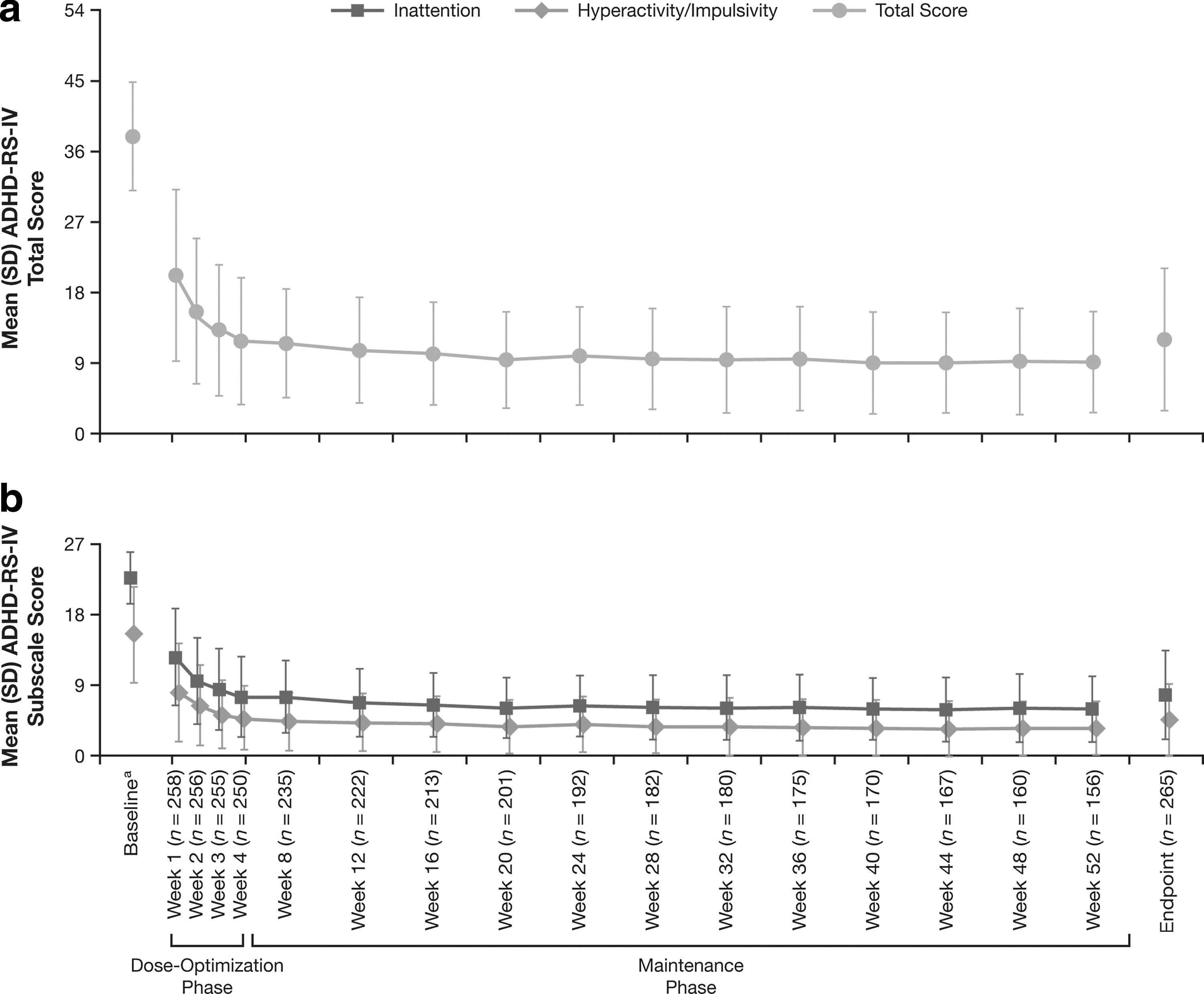
ADHD-RS-IV
A majority of the overall population was improved (CGI-I [secondary effectiveness measure] rating of 1 or 2) with LDX treatment as indicated at week 4 (228 of 250 [91.2%] participants) of the dose-optimization phase and at week 28 (178 of 182 [97.8%] participants), week 52 (153 of 156 [98.1%] participants), and end point (231 of 265 [87.2%] participants) of the maintenance phase. Overall, the mean (SD) CGI-I score at end point was 1.6 (0.86). Moreover, the percentages of participants rated as improved at end point appeared to be similar regardless of treatment assignment in the antecedent study.
The YQOL-R (secondary effectiveness measure) perceptual transformed total score significantly increased from baseline of the antecedent study at end point/ET, 28 weeks, and 52 weeks (p<0.001 for each vs. baseline) (Table 4). At end point, the change from baseline of the antecedent 4 week study in mean YQOL-R perceptual transformed domain scores (self, relationship, environment, and general QOL) also indicated significantly improved QOL (p≤0.027). The mean changes from baseline at end point in YQOL-R perceptual transformed total scores were similar regardless of original group assignments.
The YQOL-R raw score is transformed to a 0–100-point scale to assist in result interpretation; higher scores indicate improvement in QOL.
Baseline is from the antecedent 4 week study.
p<0.001 based on a two-sided one-sample t test.
ET, early termination; QOL, quality of life; SD, standard deviation; YQOL-R, Youth Quality of Life Instrument-Research Version.
Discussion
This study suggests that continued use of LDX in doses of 30, 50, and 70 mg/day in adolescents with ADHD appears to demonstrate a safety profile consistent with known effects of other long-acting psychostimulants and to provide sustained effectiveness for up to 12 months of treatment. These results also indicate that the long-term safety profile and effectiveness in adolescents are consistent with those of other long-term LDX studies conducted in children and adults with ADHD (Findling et al. 2008; Weisler et al. 2009). Administration of dose-optimized LDX was effective in the management of ADHD in an adolescent population for up to 12 months (52 weeks) regardless of prior LDX treatment status from the antecedent 4 week double-blind study. Moreover, LDX treatment demonstrated effectiveness in reducing ADHD symptoms as assessed by the ADHD-RS-IV total and subscale scores, and demonstrated global symptom improvement in the majority of participants rated by the CGI-I. Overall, participants perceived improvement in QOL at end point with LDX treatment.
There have been few long-term studies that have assessed the safety and effectiveness of psychostimulants, especially of long-acting formulations, in adolescents (Hoare et al. 2005; Hammerness et al. 2009; Findling et al. 2010). Overall, these studies found that the long-term safety profile of the psychostimulant tested was consistent with findings in short-term studies in adolescents and children with the respective compounds, and no new safety signals were detected with long-term use.
Safety
Common TEAEs (≥10%) in the current study included decreased appetite (21.1%), headache (20.8%), insomnia (12.1%), and dizziness (5.3%), and were consistent with the known effects of amphetamine treatment (Ahmann et al. 2001; Biederman et al. 2002; Ambrosini et al. 2006). Similar to other studies conducted with long-acting psychostimulants (Biederman et al. 2002; Childress et al. 2009), most TEAEs were mild to moderate in severity. TEAEs that led to early termination from this study were consistent with the safety profile of other long-acting psychostimulants used in the treatment of ADHD, regardless of patient age. Psychiatric AEs, such as psychotic or manic symptoms, hallucinations, exacerbation of thought disorder, depression, and aggressive/hostile behavior related to psychostimulant use have been reported at putatively therapeutic doses in adolescents, as well as in patients with ADHD across age groups (Metadate CD [package insert] 2009; Concerta [package insert] 2010; Adderall XR [package insert] 2011; Ritalin LA [package insert] 2012). These TEAEs in adolescents, as well as in children, with ADHD generally diminish upon cessation of psychostimulant treatment or dose adjustment (Greenhill et al. 2002).
In general, stimulants are not recommended for patients with various cardiovascular problems {2098, 175, 271, 1277} because sudden death at usual doses and serious cardiovascular AEs have been reported (Daytrana prescribing information 2010; Adderall XR prescribing information 2011; Focalin XR prescribing information 2012; Vyvanse prescribing information 2012). The American Heart Association has developed guidelines to screen and monitor children and adolescents (Gutgesell et al. 1999). These recommendations include determination of family/patient medical history prior to treatment, careful follow-up/physical examination visits that should include determination of heart rate and BP, avoiding concomitant use of psychotropic drugs and other drugs that are metabolized by or inhibit the cytochrome P-450 enzyme system, as well as ECG monitoring at baseline and during chronic therapy. Results from this study were consistent with results from trials of LDX, as well as those from trials of other long-acting psychostimulants in both children and adults with ADHD. Specifically, the modest increases in SBP, DBP, pulse rate, and heart rate seen here were comparable with those in previous LDX studies conducted in children and adults with ADHD (Adler et al. 2008; Wigal et al. 2009). As reviewed by Wolraich and colleagues, psychostimulants were reported to result in slight increases in BP and pulse rate, but these increases were usually not considered clinically significant (Wolraich et al. 2007). Further, in this study, ECG findings were deemed normal or not clinically significant with LDX treatment, consistent with other LDX studies (Wilens et al. 2006; Wigal et al. 2009).
Slight decreases in z scores for weight and BMI were observed at end point from baseline with LDX treatment (Faraone et al. 2010). These data on weight decrease were consistent with findings from a study that assessed the effects of LDX treatment on growth in children with ADHD (Faraone et al. 2010) and from other studies assessing growth with other psychostimulant therapy (National Institute of Mental Health 2004; Ambrosini et al. 2006; Spencer et al. 2006) and with other LDX studies (Findling et al. 2008; Weisler et al. 2009; Wigal et al. 2009). Although height did not change based on z scores in the present study, growth should still be monitored during treatment with stimulants, and if there is an observed slowing of growth, then the treatment regimen may need to be reevaluated.
The number of study completers (58%) in the present 52 week trial of LDX was consistent with other adolescent ADHD studies. In an open-label, multicenter extension study, in which the adolescent participants were treated with a long-acting psychostimulant, methylphenidate transdermal system, 54% of participants completed at 6 months (Findling et al. 2010). In a similar study of participants on 6 months of open-label treatment with mixed amphetamine salts extended-release formulation, 76% completed the study.
Effectiveness
Although long-term studies of psychostimulants in adolescents with ADHD are limited, the effectiveness of LDX in this population was consistent with that of other studies of psychostimulants in this age group. One large open-label study that examined the effectiveness (based on ADHD-RS-IV scores and CGI-I ratings) of mixed amphetamine salts extended release, when administered for up to 6 months in adolescents with ADHD, indicated sustained symptom improvement (Spencer et al. 2005). The majority of participants (60.9%) were rated improved at end point. In the current analysis, most participants (87.2% at end point/ET and 98.1% up to 52 weeks) were rated as improved with LDX treatment per CGI-I ratings.
QOL studies assessing psychostimulant treatment in adolescent populations with ADHD are limited. The YQOL-R results indicated significantly improved transformed scores from baseline at end point for all QOL perceptual domains. In the current study, mean (SD) YQOL-R perceptual transformed total scores at baseline were 79.8 (11.28) and at end point were 83.9 (11.00) for LDX. It should be noted that validation of the YQOL-R was conducted in adolescents with a variety of disabilities, inclusive of, but not limited to, ADHD, and indicated mean total scores of 75.2 and 82.2 for the ADHD subgroup and the normative control group, respectively (Patrick et al. 2002). Despite the relatively small increase of ∼4 units on the YQOL-R in the current study, the perceptual transformed scores at end point after treatment with LDX were similar to those of the control group of the YQOL-R validation study (Patrick et al. 2002).
In the antecedent 4 week double-blind study of LDX in adolescents, QOL at end point was not significantly improved when participants who were treated with LDX were compared with those who received placebo. That result was possibly because of the short duration of the double-blind study. The current extension study (52 weeks) indicated that QOL was improved at 28 weeks, 52 weeks, and end point/ET suggesting that a longer duration of time may be required for participants to perceive QOL improvement.
Limitations
One limitation is that the study lacked a placebo group to compare treatment effects with LDX-treated participants. The changes that occurred may be based, to some degree, on nonspecific or environmental factors experienced by the study groups, and not necessarily LDX therapy. Subjective bias based on the open-label design because of lack of blinding by investigator and study participants cannot be excluded. The baseline demographics also indicated that the participant majority was male, white, and non-Hispanic. The current data may not necessarily apply to minority adolescent populations and females; drawing conclusions should be done with caution. Exclusion in the 4 week double-blind study of participants who failed to respond to amphetamine therapy and/or were well controlled on current ADHD medication may overestimate effectiveness in the general clinical population. Participants with comorbid psychiatric disorders and/or other medical symptoms or conditions that may have contraindicated treatment with LDX were not enrolled in the current analysis. This limits the ability to generalize these findings to adolescents who have a comorbid disorder, which may be common in individuals with ADHD (Young 2008). When interpreting the decrease in ADHD symptom scores (ADHD-RS-IV scores) by week, it should be noted that mean change is based on those participants continuing in the study and, therefore, may be skewed to greater decreases, as it is possible that those discontinuing the study had poorer responses to treatment. However, two factors suggest that this may not be a prominent effect: 1) The number of participants who withdrew from the study because of lack of effectiveness was relatively modest (5/269); and 2) The differences were small between ADHD-RS-IV scores that only considered data from participants who continued to the later weeks of the study (i.e., weeks 36–52), and the end point scores of all participants (which incorporated early termination scores for those participants who were discontinued prior to completion). Another study limitation is that QOL at study entry was relatively well perceived prior to treatment and results may not represent adolescents with a lower perceived QOL.
Conclusions
This study describes the long-term safety and effectiveness of LDX treatment in a sizable adolescent population with ADHD. These results are consistent with those of prior long-term LDX studies conducted in both children and adults. Overall, the long-term use of LDX in adolescents demonstrates a safety profile consistent with that of long-acting psychostimulant use. In addition, LDX exhibits continued effectiveness for up to 52 weeks in improving core ADHD symptoms in adolescents with ADHD, similar to that in both children and adult LDX studies. Specifically, dose-optimized LDX-treated participants demonstrated ADHD symptom improvement and global impressions of clinical improvement relative to baseline of the antecedent 4 week study. Moreover, adolescents with ADHD treated with LDX also showed improved participant-perceived QOL at week 28, week 52, and end point from baseline.
Clinical Significance
As children mature into adolescence, ADHD symptoms may change, and functional impairments may result in negative impact on multiple life domains. Efficacy and safety data, as they pertain to adolescents, are important both when considering the management of ADHD and when developing additional therapeutic options in this understudied population. The results of this study of LDX safety and effectiveness over 52 weeks of open-label treatment provide clinicians with information about a new therapeutic option for adolescents with ADHD. The focus on QOL treatment outcomes offers an additional important and clinically relevant perspective.
Footnotes
Acknowledgments
Under the direction of the authors, Dr. Huda Abdullah, a former employee of SCI Communications & Information (SCI), and Dr. Michael Pucci, an employee of SCI, provided writing assistance. Editorial assistance in the form of proofreading, copy editing, and fact checking was also provided by SCI. SCI was funded by Shire Development LLC for support in writing and editing this manuscript. Drs. Thomas Babcock, Brian Scheckner, and Bryan Dirks from Shire Development LLC, also reviewed and edited the manuscript for scientific accuracy. Although the sponsor was involved in the design, collection, analysis, interpretation, and fact checking, the content, ultimate interpretation, and decision to submit the manuscript for publication to Journal of Child and Adolescent Psychopharmacology were made by the authors. The authors acknowledge the contributions to this study of Sonia Pant, Study Manager, and Diane Schneider, Medical Writer.
Disclosures
Dr. Findling receives or has received research support, acted as a consultant, received royalties from, and/or served on a speaker's bureau for Abbott, Addrenex, Alexza, American Psychiatric Press, AstraZeneca, Biovail, Bristol-Myers Squibb, Dainippon Sumitomo Pharma, Forest, GlaxoSmithKline, Guilford Press, Johns Hopkins University Press, Johnson & Johnson, KemPharm Lilly, Lundbeck, Merck, National Institutes of Health, Neuropharm, Novartis, Noven, Organon, Otsuka, Pfizer, Physicians' Post-Graduate Press, Rhodes Pharmaceuticals, Roche, Sage, Sanofi-Aventis, Schering-Plough, Seaside Therapeutics, Sepracor, Shionogi, Shire, Solvay, Stanley Medical Research Institute, Sunovion, Supernus Pharmaceuticals, Transcept Pharmaceuticals, Validus, WebMD, and Wyeth. Dr. Cutler receives or has received research support, acted as a consultant and/or speaker, and participated in a CME Advisory Board for Abbott, Addrenex, AstraZeneca, Bristol-Myers Squibb, Cephalon, GlaxoSmithKline, Janssen, Jazz, Johnson & Johnson Pharmaceutical Research & Development, LLC, Lilly, McNeil, Memory, Merck, Neuroscience Education Institute, Novartis, Ortho-McNeil, Otsuka, Pfizer, Sanofi (including Sanofi-Synthelabo, Sanofi-Aventis), Sepracor, Shionogi, Shire, Solvay, Supernus, and Targacept. Dr. Saylor receives or has received research support and acted as a consultant for Abbott, AstraZeneca, Bristol-Myers Squibb, Cephalon, Lilly, Johnson & Johnson, Merck, Novartis, Otsuka, Psychogenics, Shire, and Supernus. Dr. Gasior is an employee of Shire and holds stock and/or stock options in Shire. Mr. Hamdani is an employee of Shire and holds stock and/or stock options in Shire. Dr. Ferreira-Cornwell is an employee of Shire and holds stock and/or stock options in Shire. Dr. Childress receives or has received research support and acted as a consultant and/or speaker for Abbott, Bristol-Myers Squibb, GlaxoSmithKline, Johnson & Johnson Pharmaceutical Research & Development, LLC, Lilly USA, LLC, NextWave, Novartis, Ortho-McNeil Janssen Scientific Affairs, Pfizer, Rhodes, Sepracor, Shire, Somerset, and Sunovion.