
Abstract
Objective:
The purpose of this study was to evaluate the efficacy and safety of acute quetiapine monotherapy in adolescents with schizophrenia.
Methods:
Patients ages 13–17 years with an American Psychiatric Association, Diagnostic and Statistical Manual of Mental Disorders, 4th ed., Text Revision (DSM-IV-TR) diagnosis of schizophrenia and a Positive and Negative Syndrome Scale (PANSS) total score ≥60 were randomized to 6 weeks of quetiapine (400 or 800 mg/day) or placebo treatment. The primary efficacy measure was change in PANSS total score from baseline to day 42. Safety endpoints included adverse events and assessments of clinical chemistry values, suicidality, and extrapyramidal symptoms.
Results:
The intent-to-treat population included 220 patients. Least-squares mean change in PANSS total score from baseline to endpoint was −27.31 with quetiapine 400 mg/day, −28.44 with quetiapine 800 mg/day, and −19.15 with placebo (p=0.043 and 0.009 for quetiapine 400 and 800 mg/day, respectively, vs. placebo; mixed-model, repeated-measures analysis). Several secondary efficacy outcomes, including Clinical Global Impressions-Improvement score, supported the primary outcome measure in demonstrating significantly greater improvement in quetiapine groups than in the placebo group. Mean changes in body weight at day 42 were 2.2 kg and 1.8 kg for quetiapine 400 and 800 mg/day, respectively, and −0.4 kg for placebo. Mean changes in certain clinical chemistry parameters, including total cholesterol and triglycerides, were numerically greater in the quetiapine groups than in the placebo group. Adverse events associated with quetiapine were mostly mild to moderate in intensity and were consistent with its known profile in adults with schizophrenia.
Conclusions:
In this 6-week study of adolescent patients, quetiapine at doses of 400 and 800 mg/day provided significant improvements in symptoms associated with schizophrenia in adolescent patients, including the primary efficacy measure of PANSS total score change. Quetiapine was generally well tolerated with a profile broadly similar to that reported in adult and adolescent populations.
Clinical trial registration information:
Quetiapine Fumarate (SEROQUEL™) Compared to Placebo in the Treatment of Adolescent Patients With Schizophrenia (ANCHOR 112). Available at:
Introduction
Antipsychotics are a common choice in the treatment of adolescents with schizophrenia, particularly agents in the atypical antipsychotic class, which are associated with a reduced propensity for extrapyramidal side-effects when compared with conventional antipsychotics in this age group (Shaw and Rapaport 2006; Shaw et al. 2006; Kumra et al. 2008). Investigations of the efficacy and safety of atypical antipsychotics in adolescents with schizophrenia include, in addition to open-label studies, recent short-term, placebo-controlled trials of risperidone (Haas et al. 2009), olanzapine (Kryzhanovskaya et al. 2009), and aripiprazole (Findling et al. 2008). These trials report significant efficacy for these atypical antipsychotics compared with placebo.
Quetiapine is an atypical antipsychotic approved by the U.S. Food and Drug Administration (FDA) for the treatment of adults with schizophrenia, acute manic episodes associated with bipolar I disorder, depressive episodes associated with bipolar disorder, and the maintenance treatment of bipolar I disorder (as adjunct therapy to lithium or divalproex). An extended-release formulation of quetiapine is also approved for the adjunctive treatment of major depressive disorder.
Evidence that quetiapine may also offer efficacy in adolescents with schizophrenia derives from three open-label studies, between 3–12 weeks' duration, that investigated quetiapine at doses up to 800 mg/day (McConville et al. 2000; Shaw et al. 2001; Schimmelmann et al. 2007). McConville et al. (2000) reported a 23-day study of 10 patients ages 12–16 years with a diagnosis of schizoaffective disorder or bipolar disorder with psychotic features. Quetiapine at doses up to 800 mg/day was associated with significant improvement in psychotic symptoms, measured by change in the Brief Psychiatric Rating Scale (BPRS) total score. Shaw et al. (2001) described an 8-week study of 15 patients ages 13–17 years with schizophrenia, schizoaffective disorder, bipolar disorder, psychosis not otherwise specified, or major depressive disorder. Quetiapine at doses between 300 mg/day and 800 mg/day significantly reduced psychotic symptoms measured by rating scales including the Positive and Negative Syndrome Scale (PANSS) and the BPRS. Finally, Schimmelmann et al. (2007) investigated quetiapine at doses between 200 mg/day and 800 mg/day for 12 weeks in 56 patients ages 12–17 years diagnosed with schizophrenia, schizophreniform, or schizoaffective disorders. Quetiapine was associated with significant improvements in PANSS total and positive scores, and demonstrated a safety profile in line with previous studies in this age group.
The current 6-week, double-blind, randomized, placebo-controlled trial (D1441C00112) evaluated the efficacy and safety of quetiapine at fixed doses of 400 mg/day or 800 mg/day for the acute treatment of adolescents with schizophrenia. Based in part on the results of the current study, the immediate-release formulation of quetiapine has been approved by the FDA for the treatment of schizophrenia in adolescents ages 13–17 years. Quetiapine is also approved for the acute treatment of manic episodes of bipolar disorder in children and adolescents ages 10–17 years.
Methods
Study design
This double-blind, randomized, placebo-controlled, parallel-group study of quetiapine or placebo monotherapy in adolescents was conducted at 43 centers in Asia, Central and Eastern Europe, South Africa, and the United States between October 2004 and June 2007.
After a medication washout period ranging from 1 to 28 days (depending upon the medications involved and at the discretion of the investigator), patients with a diagnosis of schizophrenia were randomized to receive quetiapine 400 mg/day, quetiapine 800 mg/day, or placebo, for 6 weeks. If the washout period was ≥14 days, baseline safety assessments were repeated at the end of the washout period.
The study was approved by institutional review boards at each site and was performed in accordance with the current amendment of the Declaration of Helsinki and International Conference on Harmonization/Good Clinical Practice guidelines. All patients and their legal guardians provided written, informed assent and consent, respectively, before study participation. Patients could withdraw from the study at any time if they or their legal guardian no longer wished to participate. Patients could also be discontinued from the study at any time at the discretion of the investigator.
Patient population
Male and female inpatients or outpatients ages 13–17 years with a diagnosis of schizophrenia based on the American Psychiatric Association, Diagnostic and Statistical Manual of Mental Disorders, 4th Edition, Text Revision (DSM-IV-TR) (American Psychiatric Association 2000) were eligible for inclusion in the study. The diagnosis of schizophrenia was confirmed by administering the Kiddie Schedule for Affective Disorders and Schizophrenia for School-Age Children—Present and Lifetime Version (K-SADS-PL) (Kaufman et al. 1997). Patients recruited to the study were required to be symptomatic at enrollment, although they could be receiving treatment with an antipsychotic medication. Patients were required, at screening and randomization, to have a PANSS total score ≥60 and a score ≥4 on at least one of the following PANSS items: delusions (P1), conceptual disorganization (P2), or hallucinations (P3), which corresponds to at least moderate symptomatology. Clinicians (e.g., child psychiatrists or psychologists) trained in the use of the rating scales administered the scales.
Patients were excluded from the study if they had a DSM-IV Axis I diagnosis of bipolar disorder, schizophreniform disorder, schizoaffective disorder, psychotic disorder not otherwise specified, or acute posttraumatic stress disorder. Patients with psychosis judged to be a direct consequence of a medical condition or its treatment were also excluded. Other exclusion criteria included a history of suicide attempts or homicidal risk or behavior within the past 3 months, as judged by the investigator; DSM-IV-defined substance abuse or dependence; laboratory test results outside the normal reference range; and a hospital admission for diabetes or diabetes-related illness in the past 3 months. Renal, cardiovascular, hepatic, hematologic, endocrinologic, ophthalmologic, or other medical conditions that were unstable or may have affected or been affected by the study medication led to exclusion. Pregnancy and lactation were additional exclusion criteria.
Random assignment to treatment was achieved by a central randomization service, with stratification by gender. Patients were assigned to one of the three groups: quetiapine 400 mg/day, quetiapine 800 mg/day, or placebo in a 1:1:1 ratio. The distribution of treatments was balanced within study regions (i.e., Asia, Central and Eastern Europe, South Africa, and the United States).
Study medication
The two fixed doses of quetiapine investigated in this study (400 mg/day and 800 mg/day) were selected based on the experience of open-label studies of the efficacy and safety of quetiapine in adolescents (McConville et al. 2000; Shaw et al. 2001) and from the effective doses observed in the treatment of adults with schizophrenia (Small et al. 1997; Zhong et al. 2006). Quetiapine was initiated at a dose of 50 mg on day 1, 100 mg on day 2, and then titrated in 100 mg/day increments in divided doses to achieve the target dose of 400 mg/day by day 5 or 800 mg/day by day 9. Quetiapine or placebo was administered orally twice daily, with an option for administration three times daily at the discretion of the investigator based on the observed tolerability profile. The quetiapine and placebo tablets were identical in size and color in order to maintain blinding.
Concomitant medication
Continued use of selected antidepressants (citalopram, escitalopram, sertraline, bupropion, or venlafaxine) was permitted if the dose had been stabilized at least 30 days before enrollment. Ongoing treatment with lorazepam (or equivalent benzodiazepine) at a dose up to 4 mg/day was also permitted, but could not exceed 4 days in any study week. Use of antipsychotics, psychostimulants, CYP 3A4 inhibitors/inducers, monoamine oxidase inhibitors, atomoxetine, and antidepressants other than those noted was prohibited during the study period.
Efficacy evaluations
The primary efficacy measure was the mean change from baseline to day 42 in the 30 item PANSS total score (Kay et al. 1987). Secondary efficacy measures included changes in PANSS subscale scores and PANSS-derived Brief Psychiatric Rating Scale (BPRSd) (Overall and Gorham 1962) and the proportions of patients who achieved a response (defined as a ≥30% reduction in the PANSS total score from baseline). Additional secondary efficacy measures included score changes on the Clinical Global Impressions-Severity of Illness scale (CGI-S) (Guy and Bonato 1970), CGI Improvement scale (GCI-I) (Guy and Bonato 1970), Children's Global Assessment Scale (CGAS) (Shaffer et al. 1983), and Caregiver Strain Questionnaire (CGSQ) (Brannan et al. 1997), based on a new scoring convention, range 0–15, which calculates a global score from the sums of subscale scores: Internalized subjective strain, externalized subjective strain, and objective strain. Higher scores are indicative of greater degrees of caregiver strain.
Assessments of PANSS and CGI-S scores were made at baseline (day 1) and weekly from day 7 to day 42. CGAS and CGSQ scores were assessed at baseline and on day 42. CGI-I was additionally assessed at day 42.
Safety and tolerability evaluations
Safety and tolerability assessments included the reported incidence and severity of adverse events and withdrawals related to adverse events. Adverse events were recorded on adverse event forms and classified according to the Medical Dictionary for Regulatory Activities (MedDRA). Adverse events were rated as medication related based on investigator judgment. A serious adverse event included an event that resulted in death or was immediately life-threatening, required hospitalization or prolongation of existing hospitalization, resulted in persistent or significant disability or incapacity, may have jeopardized the patient, or required medical intervention to prevent one of these outcomes.
Clinical and laboratory parameters included vital signs; 12-lead electrocardiogram (ECG) variables; lipid panel; glucose concentration; glycosylated hemoglobin (HbA1c); thyroid, liver, and renal function; and prolactin concentration. Laboratory values were determined at a central laboratory. The incidences of potentially clinically significant shifts in clinical chemistry values, hematology values, and vital signs and ECG parameters were explored based on criteria that are defined in Appendix Tables A1, A2, and A3. Extrapyramidal symptoms (EPS) were assessed by reported adverse event rates, the Simpson-Angus Rating Scale (SAS) (Simpson and Angus 1970), the Barnes Akathisia Rating Scale (BARS) (Barnes 1989), the sum of the first seven individual items on the Abnormal Involuntary Movement Scale (AIMS-7) (Guy 1976), and use of the anticholinergic, benztropine, to treat emergent EPS (prophylactic use of benztropine was not permitted).
Adverse events, weight, vital signs, and EPS scores were assessed at baseline and weekly from day 7 to day 42. Clinical chemistry and hematology parameters were assessed at the screening visit and on day 42 or the final visit, whereas glucose was additionally assessed on day 28. A toxicology screen by urinalysis was performed at baseline and at final visit to identify substance use.
Statistical analyses
Efficacy analyses were performed on the intent-to-treat (ITT) population, which included all patients who had had a baseline and at least one set of postbaseline assessments. The primary efficacy measure was the mean change in the PANSS total score from baseline to day 42, analyzed using mixed-model, repeated-measures (MMRM) analysis, with baseline PANSS score as a covariate and treatment, study region, visit, and visit-by-treatment interaction as fixed effects.
For secondary efficacy measures, MMRM was used to analyze changes in PANSS subscales and CGI-S scores, generalized estimating equations (GEE) assessed response rate and CGI-I scores, and analysis of covariance (ANCOVA) was used to assess changes in CGAS and CGSQ scores.
Statistical tests for treatment differences were performed using a two-sided hypothesis test. Where appropriate, 95% confidence intervals (CI) were also calculated. The primary analysis employed a Simes–Hommel step-up procedure to adjust for the two comparisons (quetiapine 400 mg/day and 800 mg/day) with placebo. Secondary analyses reported nominal 5% levels of significance.
To test for homogeneity of treatment effects, the primary analysis was repeated on the per-protocol (PP) population, which excluded patients with protocol violations or deviations. Analyses using ANCOVA with last-observation-carried-forward (LOCF) imputation were also conducted to support the primary MMRM analysis.
Safety and tolerability analyses were conducted on the safety population, which included all patients who took at least one dose of study medication. Descriptive statistics were used to characterize adverse events and changes in clinical and laboratory parameters. ANCOVA was used to compare changes in prolactin concentration in the quetiapine and placebo groups. Suicidality analyses were conducted post hoc utilizing standardized classifications similar to those in the Columbia Suicidality Classification Project (Posner et al. 2007). Categorization of SAS and AIMS-7 score changes was analyzed by GEE.
Sample sizes were determined to provide at least 85% power to detect a difference of 15 points between the quetiapine 400 mg/day or 800 mg/day group and the placebo group with respect to the primary efficacy analysis (i.e., mean change from baseline in PANSS total score).
Results
Patients and disposition
Of 268 patients screened, 222 patients were randomly assigned to receive quetiapine 400 mg/day, quetiapine 800 mg/day, or placebo (Fig. 1). The primary reason for screen failure was failure to fulfill eligibility criteria. Two serious adverse events (worsening of schizophrenia, gastritis) occurred in two patients during washout; the case of worsening of schizophrenia was considered secondary to washout and the patient was not randomized, whereas the patient with gastritis completed the study. All 222 randomly assigned patients received at least one dose of study medication and were included in the safety population. Of these, 220 patients additionally had at least one postbaseline assessment and were included in the ITT population (n=73, quetiapine 400 mg/day; n=74, quetiapine 800 mg/day; n=73, placebo). The PP population included 171 patients at randomization (n=56, quetiapine 400 mg/day; n=61, quetiapine 800 mg/day; n=54, placebo). A total of 164 patients completed the study protocol (n=56, 61, and 47, respectively). Patients were excluded from the PP population at similar rates in the three treatment groups; a common reason for discontinuation in all groups related to use of prohibited anxiolytics or hypnotics (n=10, quetiapine 400 mg/day; n=8, quetiapine 800 mg/day; n=15, placebo).

Disposition of adolescents with schizophrenia in the screening and randomized treatment phases for the quetiapine 400 mg/day, quetiapine 800 mg/day, and placebo groups. Study-specific discontinuation criteria include severe noncompliance (as judged by the investigator), symptom deterioration, or inability to tolerate the assigned dose.
Demographic and baseline disease characteristics were similar in the three treatment groups (Table 1 and Appendix Table A4). The mean age of patients was 15.4 years and ∼41% were female. The most common diagnostic subtype in all groups was paranoid schizophrenia, representing 70.5% of all patients. Use of antipsychotics during the 30 days preceding the study was reported in 60.3%, 60.8%, and 64.0% of patients in the quetiapine 400 mg/day, quetiapine 800 mg/day, and placebo group, respectively. Prior use of antidepressants was higher in the 400 mg/day quetiapine group (11.0%) than in the 800 mg/day (1.4%) or placebo (2.7%) groups. Prior use of anxiolytics/hypnotics was broadly similar in the respective groups (30.2%, 24.4%, and 27.9%), whereas prior use of mood stabilizers or psychostimulants was low overall (5.9% and 2.3% respectively, all groups combined). The incidence of substance use during the study was 9.6% in the quetiapine 400 mg/day, 6.8% in the quetiapine 800 mg/day, and 16.0% in the placebo groups.
ITT=intent-to-treat; SD=standard deviation; BMI=body mass index; DSM-IV=Diagnostic and Statistical Manual of Mental Disorders, 4th edition; PANSS=Positive and Negative Syndrome Scale.
Rates of study completion were higher in the two quetiapine dose groups (76.7% in the quetiapine 400 mg/day group and 82.4% in the quetiapine 800 mg/day group) than in the placebo group (62.7%). The most common reasons for study withdrawal were the development of study-specific discontinuation criteria (8.2% in the quetiapine 400 mg/day group and 20.0% in the placebo group) and the development of adverse events (9.5% in the quetiapine 800 mg/day group) (Fig. 1). Study-specific discontinuation criteria included withdrawal of informed consent, severe noncompliance as judged by the investigator, symptom deterioration, or inability to tolerate the assigned dose. Adverse events associated with withdrawal included neutropenia, somnolence, anxiety, elevated mood, and schizophrenia in the quetiapine 400 mg/day group (five patients); nausea, fatigue, rubella, dysarthria, sedation, somnolence, depression, suicidal ideation, and dyspnea in the quetiapine 800 mg/day group (seven patients); and delusion and schizophrenia in the placebo group (two patients).
Rates of treatment compliance, assessed by returned-tablet count, were high in all groups: 97.3% in the quetiapine 400 mg/day group, 96.1% in the quetiapine 800 mg/day group, and 98.7% in the placebo group. Concomitant antidepressant use was generally low across groups at 6.9%, 1.4%, and 2.7%, respectively, and lorazepam (or equivalent) was used by 27.4%, 17.6%, and 25.3%, respectively. Anticholinergic medication for the treatment of emergent EPS was used by 5.5% in the quetiapine 400 mg/day group, 1.4% in the quetiapine 800 mg/day group, and 0.0% in the placebo group.
Efficacy
PANSS-based analyses: PANSS total score change
At day 42, patients in the quetiapine 400 mg/day and quetiapine 800 mg/day groups demonstrated significantly greater mean improvements in the PANSS total score compared with placebo-treated patients in the ITT population (the primary efficacy evaluation). Least-squares mean changes in PANSS total scores by MMRM analysis were −27.31 (standard error [SE]: 2.64) with quetiapine 400 mg/day, −28.44 (SE: 1.82) with quetiapine 800 mg/day, and −19.15 (SE: 3.04) with placebo (Table 2). Mean differences from placebo at day 42 were −8.16 (SE: 4.01; p=0.043) with quetiapine 400 mg/day and −9.29 (SE: 3.52; p=0.009) with quetiapine 800 mg/day. Significant divergence from placebo in the PANSS score change was first observed at day 14 in the quetiapine 800 mg/day group (p=0.012) and at day 21 in the quetiapine 400 mg/day group (p=0.006) (Fig. 2). Significant divergence from placebo was maintained at all assessments in the quetiapine 800 mg/day group from day 14 until study end.
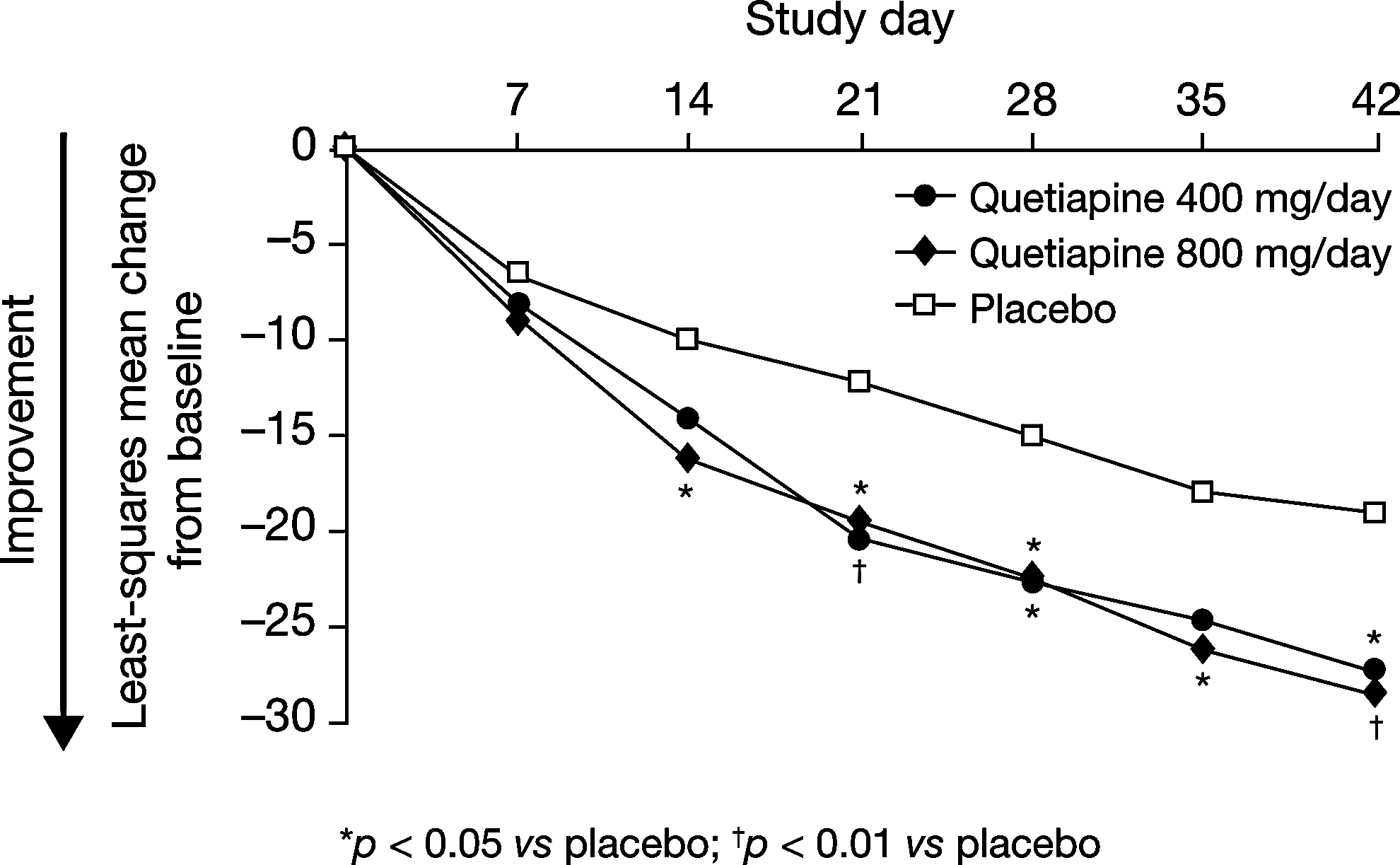
Least-squares mean change in Positive and Negative Syndrome Scale (PANSS) total score from baseline to day 42 in adolescents treated with quetiapine 400 mg/day, quetiapine 800 mg/day, or placebo (mixed-model, repeated-measures [MMRM], observed cases, intent-to-treat [ITT] population).
PANSS=Positive and Negative Syndrome Scale; ITT=intent-to-treat; MMRM=mixed-model, repeated-measures; SE=standard error; CGI=Clinical Global Impressions; CGAS=Children's Global Assessment Scale; ANCOVA=analysis of covariance; BPRSd=PANSS-derived Brief Psychiatric Rating Scale; CGSQ=Caregiver Strain Questionnaire.
PANSS total score changes from baseline to day 42 in the ITT population were further analyzed by gender, region, study completion versus withdrawal status, pattern of missing data, and previous quetiapine exposure. No significant treatment interactions were observed in these analyses. In patients who were hospitalized at baseline (43.2% overall; Table 1), mean (SD) PANSS total score changes were −20.9 (27.53) with quetiapine 400 mg/day, −27.9 (16.12) with quetiapine 800 mg/day, and −12.9 (25.72) with placebo. In patients not hospitalized at baseline, respective PANSS total score changes were −26.3 (24.72), −24.9 (19.62), and −20.5 (25.65).
ANCOVA analysis with LOCF imputation supported MMRM analysis for the primary endpoint in the ITT population. Least-squares mean changes in PANSS total score at day 42 by ANCOVA/LOCF analysis were −25.76 (SE: 2.47) with quetiapine 400 mg/day, −27.23 (SE: 2.45) with quetiapine 800 mg/day, and −18.52 (SE: 2.50) with placebo, representing significant divergences from placebo with both quetiapine 400 mg/day (−7.24, SE: 3.44; p=0.036) and quetiapine 800 mg/day (−8.71, SE: 3.42; p=0.012). Similar to MMRM analysis, ANCOVA/LOCF analysis of PANSS total score change demonstrated first occurrence of significant divergence from placebo at day 14 in the quetiapine 800 mg/day group (p=0.028) and at day 21 in the quetiapine 400 mg/day group (p=0.014).
In the PP population, least-squares mean decreases in PANSS total score at day 42 relative to placebo were −5.49 (SE: 4.38) with quetiapine 400 mg/day and −6.72 (SE: 3.93) with quetiapine 800 mg/day, based on MMRM analysis. These differences did not attain statistical significance (p=0.212 and 0.090, respectively).
Response rate
Using LOCF imputation, the proportion of patients who achieved the a priori defined response criterion (≥30% reduction in the PANSS total score at day 42) was 38.4%, 36.5%, and 26.0% in the quetiapine 400 mg/day, quetiapine 800 mg/day, and placebo groups, respectively (p=0.109 with quetiapine 400 mg/day and p=0.194 with quetiapine 800 mg/day vs. placebo). Using observed cases data, response rates were 51.9% in the quetiapine 400 mg/day group, 40.0% in the quetiapine 800 mg/day group, and 39.5% in the placebo group. GEE analysis indicated no significant difference in the odds of response between the quetiapine groups and the placebo group (p=0.125 with quetiapine 400 mg/day and p=0.675 with quetiapine 800 mg/day vs. placebo).
PANSS subscale score changes
Improvements in least-squares mean PANSS positive symptom subscale scores at day 42 were significantly greater with quetiapine 800 mg/day relative to placebo (−2.83; SE: 1.05; p=0.008) (Table 2). Improvements in least-squares mean sum scores for PANSS items P4, P7, G8, and G14 (PANSS aggression/hostility cluster) at day 42 relative to placebo were significantly greater with both quetiapine 400 mg/day (−1.76; SE: 0.74; p=0.018) and quetiapine 800 mg/day (−1.99; SE: 0.69; p=0.005) (Table 2).
Improvements in the BPRSd total score were significant in the quetiapine 800 mg/day group (p=0.010). Other, nonsignificant, changes in PANSS subscale scores are described in Table 2.
CGI-S and CGI-I
Least-squares mean differences in CGI-S scores were significant between the quetiapine 800 mg/day and placebo groups (−0.47; SE: 0.20; p=0.018) (Table 2). Mean CGI-I scores also indicated significantly greater improvements in quetiapine- than in placebo-treated groups. Least-squares mean differences from placebo at day 42 were −0.60 (SE: 0.24; p=0.013) with quetiapine 400 mg/day and −0.77 (SE: 0.23; p<0.001) with quetiapine 800 mg/day (Table 2).
The proportion of patients “much improved” or “very much improved” on the CGI-I scale at day 42 was 60.0% in the quetiapine 400 mg/day group and 56.4% in the quetiapine 800 mg/day group compared with 41.9% in the placebo group using observed cases data. GEE analysis indicated significantly higher odds of improvement with both quetiapine 400 mg/day and quetiapine 800 mg/day than with placebo (odds ratio 2.81 [p=0.009] and 2.71 [p=0.014], respectively). Using LOCF imputation, the proportions of patients “much improved” or “very much improved” were 49.3%, 52.7%, and 27.4% in the quetiapine 400 mg/day, quetiapine 800 mg/day, and placebo groups, respectively.
CGAS
Mean CGAS total score changes were statistically significantly greater with quetiapine 800 mg/day than with placebo. At day 42, the least-squares mean difference from placebo by ANCOVA analysis was 5.05 (SE: 2.13; p=0.019) (Table 2).
CGSQ
CGSQ total score changes at day 42 indicated a statistically significantly lower overall caregiver burden in patients treated with quetiapine 400 mg/day. At day 42, the least-squares mean difference from placebo by ANCOVA analysis was −0.86 (SE: 0.32; p=0.008) (Table 2).
Safety and Tolerability
Adverse events
Overall incidences of adverse events in the quetiapine 400 mg/day, quetiapine 800 mg/day, and placebo groups were 79.5%, 74.3%, and 60.0%, respectively, with medication-related adverse events reported in 56.2%, 46.0%, and 22.7%, respectively. The most common adverse events associated with quetiapine included somnolence, headache, and dizziness (Table 3). Most adverse events were mild to moderate in intensity. Treatment discontinuations because of adverse events occurred in 6.9%, 9.5%, and 2.7% of patients receiving quetiapine 400 mg/day, quetiapine 800 mg/day, and placebo, respectively, and sedation was one of the most frequent causes of discontinuation.
Rates of serious adverse events were 5.5% (n=4), 6.8% (n=5), and 5.3% (n=4) of patients in the quetiapine 400 mg/day, quetiapine 800 mg/day, and placebo groups, respectively. The majority of serious adverse events were psychiatric disorders, which were observed in three patients in the quetiapine 400 mg/day group (visual hallucination, psychotic disorder, and schizophrenia), three patients in the quetiapine 800 mg/day group (aggression, agitation and restlessness, and schizophrenia), and three patients treated with placebo (aggression and insomnia, delusion, and schizophrenia).
Weight change
In this population who have not yet reached their full adult height and weight, mean body weight at baseline was 61.0 kg in the quetiapine 400 mg/day group, 61.7 kg in the quetiapine 800 mg/day group, and 62.5 kg in the placebo group (Table 1). Mean (SD) change in body weight from baseline to day 42 was 2.2 (2.6) kg, 1.8 (2.8) kg, and −0.4 (3.5) kg, respectively, and rates of weight gain ≥7% were 23.2%, 18.2%, and 6.8%, respectively. Changes in mean (SD) age- and gender-adjusted body mass index (BMI) z-score from baseline to day 42 in the quetiapine 400 mg/day, quetiapine 800 mg/day, and placebo group were 0.26 (0.39), 0.20 (0.46), and −0.01 (0.29), respectively. When patients were grouped into categories of healthy weight, at-risk of overweight, and overweight at baseline, following the U.S. Centers for Disease Control and Prevention BMI criteria, low numbers of patients were observed to change their BMI category during the study period (Appendix Table A5).
Clinical chemistry parameters
Changes from baseline to final visit in selected clinical chemistry parameters are shown in Table 4. Mean changes in the lipid panel included increases in total cholesterol, low-density lipoprotein (LDL) cholesterol, and triglycerides in both quetiapine groups compared with decreases in these parameters in the placebo group. Mean changes in high-density lipoprotein (HDL) cholesterol were similar in the three treatment groups.
The protocol specified fasting blood draws (fasting was defined as patient reported ≥8 hours between time of last meal and time of blood draw). Not all patients in this study were confirmed fasting.
HbA1c=glycated hemoglobin.
Mean changes from baseline in glucose and HbA1c levels under fasting conditions (i.e., measured at least 8 hours since the last meal) were similar in the three treatment groups. On day 7, one patient in the quetiapine 800 mg/day group reported an adverse event potentially associated with diabetes; this event was not considered by the investigator to be related to study medication. In four patients who had one or more of the following findings (i.e., documented fasting glucose ≥126 mg/dL at randomization, undocumented fasting glucose ≥200 at baseline, HbA1c above the upper limit of normal at randomization, or a history of diabetes), and in 18 additional patients judged to have risk factors for the development of diabetes (i.e., fasting glucose ≥100 and <126 mg/dL at randomization, history of gestational diabetes, or BMI≥35 kg/m2), there were no changes in glucose or insulin values that were considered to be clinically significant.
Mean changes at final visit in parameters assessing liver function (aspartate transaminase and alanine aminotransferase) and renal function (creatinine and blood urea nitrogen) were minimal and similar in the three treatment groups. Mean (SD) prolactin concentrations at baseline were higher in the placebo group (28.7 [29.13] ng/mL) compared with the quetiapine 400 mg/day and 800 mg/day groups (20.8 [17.01] and 18.1 [20.09] ng/mL, respectively). ANCOVA performed on log transformed data to correct the baseline skew in distribution of values indicated that least-squares mean prolactin values at day 42 were similar in the three groups (7.29, 7.07, and 7.47 ng/mL, respectively), possibly reflecting the short washout period of previous antipsychotic therapy. Mean changes in prolactin levels in male and female subgroups are shown in Table 4. Mean total thyroxine levels decreased in the two quetiapine treatment groups, accompanied by a mean increase in thyroid stimulating hormone in the quetiapine 400 mg/day group (Table 4).
The incidences of potentially clinically significant shifts in clinical chemistry parameters in the three treatment groups are shown in Appendix Table A1.
Hematology values
Changes in hematology values and adverse event reports associated with abnormal hematology values indicated no clinically significant differences among the three treatment groups. The incidences of potentially clinically significant shifts in hematology parameters in the three treatment groups are shown in Appendix Table A2.
Vital signs and ECG variables
Mean changes in blood pressure (systolic and diastolic, supine and standing) from baseline to final assessment were similar in the three treatment groups. For example, mean (SD) changes in standing diastolic blood pressure at final visit were 2.1 (8.65), 1.1 (10.24), and −1.2 mm Hg (7.68), respectively, in the quetiapine 400 mg/day, quetiapine 800 mg/day, and placebo groups. Mean changes in pulse rate were greater in the quetiapine groups than in the placebo group at final visit. For example, mean (SD) changes in standing pulse rate were 6.3 (13.12) bpm, 2.2 (17.08) bpm, and −2.5 (13.14) bpm in the respective groups.
Two events of fainting, potentially associated with syncope, were reported for one patient in the quetiapine 400 mg/day group. Both events were moderate in intensity and did not result in discontinuation. The incidences of potentially clinically significant shifts in vital signs and ECG variables are shown in Appendix Table A3.
EPS
Adverse events potentially associated with EPS (including akathisia, tremor, extrapyramidal disorder, hypokinesia, restlessness, psychomotor hyperactivity, muscle rigidity, and dyskinesia) occurred in 12.3%, 13.5%, and 5.3% of patients in the quetiapine 400 mg/day, quetiapine 800 mg/day, and placebo groups, respectively. The majority of these adverse events were rated mild to moderate in intensity, and none resulted in study withdrawal. Changes in SAS, AIMS, and BARS scores from baseline to final assessment at day 42 were small and similar in all treatment groups. The odds of a worsening in movement disorders at day 42 were not significantly different in the quetiapine groups compared with the placebo group, as assessed by GEE analysis of SAS scores (p=0.264 and 0.158 with quetiapine 400 mg/day and quetiapine 800 mg/day, respectively) and AIMS-7 scores (p=0.143 and 0.839, respectively).
Suicidality
No patient completed suicide during this study. A serious adverse event potentially related to suicidality was reported in two (2.7%) patients in the quetiapine 800 mg/day group (intentional self-injury and suicidal ideation, respectively) and in one (1.4%) patient in the placebo group (intentional self-injury). These three patients were also identified in the post-hoc analysis using the Columbia Suicidality Classification scale (CSCS), with classifications of self-injurious behavior (two patients) and suicidal ideation (one patient). CSCS additionally identified one patient in the quetiapine 800 mg/day group with suicidal ideation (who experienced a moderate worsening of schizophrenia symptoms) and one patient in the placebo group with a possibly suicidal event (a mild contusion hematoma in the right zygomaticotemporal region).
Discussion
This is the first large, randomized, placebo-controlled trial to investigate the efficacy and safety of quetiapine as monotherapy in the acute treatment of adolescents with schizophrenia. Quetiapine monotherapy for 6 weeks at a fixed dose of 400 mg/day or 800 mg/day was associated with significantly greater improvements in PANSS total score (the primary efficacy measure) compared with placebo. The magnitude of the PANSS score divergence from placebo at day 42 (mean 8.16 points with quetiapine 400 mg/day and 9.29 points with quetiapine 800 mg/day by MMRM analysis) indicates a clinically meaningful effect for quetiapine therapy in this patient population, which experienced, on average, moderate to severe illness at baseline. Significant divergence from placebo in PANSS total score was achieved at day 14 in the quetiapine 800 mg/day group and at day 21 in the quetiapine 400 mg/day group, suggesting an early and potentially dose-dependent onset of symptom improvement.
Quetiapine-treated patients additionally experienced significant improvements in some, but not all, secondary efficacy measures, in support of the primary efficacy outcomes. Although not directly comparable in terms of methodology or the diagnostic groups investigated, this study provided efficacy outcomes consistent with previously reported outcomes in open-label studies of adolescents (McConville et al. 2000; Shaw et al. 2001; Schimmelmann et al. 2007) and controlled studies of adults with schizoprenia (Riedel et al. 2007).
In general, the adverse event profile of quetiapine was also consistent with earlier studies of patients with schizophrenia, including adolescent populations (McConville et al. 2000; Shaw et al. 2001; Schimmelmann et al. 2007). The most commonly reported adverse events in quetiapine-treated patients were somnolence, headache, and dizziness, which in most cases were rated mild to moderate in intensity, and were associated with treatment withdrawal in a low number of patients. Rates of serious adverse events were similar in the quetiapine and placebo groups.
A number of safety parameters in this study indicated differences between quetiapine- and placebo-treated patients, similar to the observations in another study of children and adolescents ages 4–19 years (Correll et al. 2009). These included divergences in mean change in weight, blood pressure, heart rate, and laboratory measures of lipid and thyroid function. These changes in laboratory parameters are broadly consistent with the changes observed in adults with schizophrenia (Riedel et al. 2007; Seroquel® Prescribing Information, 2012). Although no clinically significant differences in laboratory parameters were noted in this study, the current recommendation is to measure fasting blood lipid levels and blood pressure in children and adolescents at the beginning of and periodically during treatment, and to monitor fasting blood glucose testing in patients with risk factors for diabetes mellitus (Seroquel® Prescribing Information, 2012). Recommendations on the timings of follow-up monitoring include assessments of fasting plasma glucose, lipid level, and blood pressure at 3 months with annual assessments thereafter, and weight assessments at 4, 8, and 12 weeks (American Diabetes Association et al. 2004).
Although no relationship between treatment and suicidal behavior/ideation was established in this study, it should be noted that the FDA has issued a boxed warning for increased risk of suicidal thinking and behavior in children, adolescents, and young adults taking antidepressants (including quetiapine) for major depressive disorder and other psychiatric disorders.
Limitations
Limitations of the current study that may impact the interpretation of outcomes include the use of fixed doses of quetiapine, which differs from the flexible dosing approach commonly used in clinical practice. Also, a study with this design cannot offer insights into the comparative effectiveness of quetiapine and alternative medications, for which purpose comparative studies have a greater role (e.g., Sikich et al. 2008). When interpreting the efficacy outcomes, it is important to consider that there was residual symptomatology in patients at study completion, as indicated by the mean PANSS scores, suggesting the potential for subsequent improvements in symptomatology. Furthermore, the meaningful benefits that were observed in the placebo group impact the degree of symptom amelioration that can be attributed to the active medication; of note, similar substantive placebo response rates were also reported in the pediatric studies of risperidone (Haas et al. 2009), olanzapine (Kryzhanovskaya et al. 2009), and aripiprazole (Findling et al. 2008). Another limitation associated with this study is that its brevity precludes statements about the long-term safety of quetiapine in this patient population. An open-label investigation of the long-term safety of quetiapine therapy that included patients who were enrolled in the current study will be the topic of a subsequent manuscript (D1441C00150). Finally, data were not available in this population to assess the impact of prior antipsychotic use on weight gain; this represents an area for future investigation.
In spite of the study limitations, this study has a role in informing clinical practice because of the large population size and the broad range of efficacy and safety measures that were investigated.
Conclusion
In this 6-week, placebo-controlled efficacy and safety study, quetiapine at a dose of 400 mg/day and 800 mg/day provided significant improvements in symptoms associated with schizophrenia in adolescent patients, including the primary efficacy measure of PANSS total score change. Quetiapine was generally well tolerated with a profile broadly similar to that reported previously in adult and adolescent populations.
Clinical Significance
This article describes the first large, randomized, placebo-controlled study of the efficacy and safety of quetiapine monotherapy in the treatment of adolescents with schizophrenia. The study demonstrated that quetiapine for 6 weeks at fixed doses of 400 or 800 mg/day was associated with significantly greater improvements in PANSS total score (the primary efficacy measure) compared with placebo. Secondary efficacy outcomes supported the primary outcome measure. Adverse events associated with quetiapine were consistent with its profile in adults with schizophrenia. The large population size that was studied and the broad range of efficacy and safety measures that were investigated support a role for this study in informing clinical practice.
Disclosures
The authors declare that this work was supported by AstraZeneca Pharmaceuticals (Study D1441C00112). Dr. Findling receives or has received research support, acted as a consultant, received royalties from, and/or served on a speaker's bureau for Abbott, Addrenex, Alexza, American Psychiatric Press, AstraZeneca, Biovail, Bristol-Myers Squibb, Forest, GlaxoSmithKline, Johns Hopkins University Press, Johnson & Johnson, KemPharm, Lilly, Lundbeck, Merck, National Institutes of Health, Neuropharm, Novartis, Noven, Organon, Otsuka, Pfizer, Physicians' Post-Graduate Press, Rhodes Pharmaceuticals, Roche, Sage, Sanofi-Aventis, Schering-Plough, Seaside Therapeutics, Sepracore, Shionogi, Shire, Solvay, Stanley Medical Research Institute, Sunovion, Supernus Pharmaceuticals, Transcept Pharmaceuticals, Validus, WebMD, and Wyeth. Dr. McKenna has no financial interests to disclose. Dr. Pathak is an employee and Dr. Early and Ms. Stankowski are former employees of AstraZeneca Pharmaceuticals LP, USA.
Footnotes
Acknowledgment
We thank Bill Wolvey from PAREXEL, who provided medical writing support funded by AstraZeneca.
The Trial 112 Study Investigators
Alexey Agarkov, Tomsk Clinical Psychiatric Hospital, Tomsk, Russia; Daisy Ann Artuz, Brokenshire Hospital, Davao City, Philippines; Sarah D. Atkinson, Finger Lakes Clinical Research, Rochester, New York; Deborah Bergen, Cientifica Inc., Newton, Kansas; Bernhard Blanz, Klinik für Kinder- und Jugendpsychiatrie, Jena, Germany; Valeriy Semenovich Bitenskiy, Odessa State Medical University, Odessa, Ukraine; Ann Childress, Center for Psychiatry and Behavioral Medicine, Inc., Las Vegas, Nevada; Miroslaw Dabkowski, Oddzial VI Psychiatrii Dzieci I Mlodziezy, Toruń, Poland; Melissa DelBello, Psychiatric Professional Services, Inc., Cincinnati, Ohio; Vladislav A. Demchenko, Kiev's City Psycho-neurological Hospital #2, Kiev, Ukraine; Vladimir Diligenski, Clinical-Hospital Center “Dr.Dragiša Mišović”, Belgrade, Serbia; Joseph Fanelli, Midwest Center for Neurobehavioral Medicine, Oakbrook, Illinois; Jörg Fegert, Klinikbereich Safranberg, Ulm, Germany; Robert Findling, University Hospitals of Cleveland, Cleveland, Ohio; John Gilliam, International Clinical Research Associates, Inc., Richmond, Virginia; Georgina Gozo-Oliver, Veterans Memeorial Medical Center, Quezon City, Philippines; Harinder Grewal, ATP Clinical Research, Inc., Costa Mesa, California; Nelson Handal, Harmonex, Dothan, Alabama; Robert Hendren, UC Davis Department of Psychiatry and Behavioral Sciences, Sacramento, California; Beate Herpertz-Dahlmann, Universitätsklinikums an der Rheinisch-Westfälischen Technischen Hochschule, Aachen, Germany; Willis Holloway, Jr., Cutting Edge Research, Oklahoma City, Oklahoma; Malgorzata Janas-Kozik, Oddzial Psychiatrii Wieku Rozwojowego Centrum Pediatrii, Sosnowiec, Poland; Gregory Kaczenski, K&S Professional Research Services, Little Rock, Arkansas; Ali Kashfi, Alamonte, Florida; Linda Keyter, Stikland Hospital, Cape Town, South Africa; Saaid Khojasteh, Saaid Khojasteh & Associates, Inc., St. Charles, Missouri; James Knutson, Eastside Therapeutic Resource, Kirkland, Washington; Irina Alexandrovna Kozlova, Mental Health Research Center, Moscow, Russia; Valeriy Krasnov, Moscow Institute of Psychiatry, Moscow, Russia; David Krefetz, CNS Research Institute, PC, Clementon, New Jersey; Aneta Lakic, Clinic for Child and Adolescent Neurology and Psychiatry, Belgrade, Serbia; Cynthia Leynes, Philippine General Hospital, Manila, Philippines; Donna Londino, Medical College of Georgia Department of Psychiatry and Health Behavior, Augusta, Georgia; Matlala Mabeba, Private Rooms, Polokwane, South Africa; Ramesh Kumar Mahendru, Mahendru Psychiatric Centre, Kanpur, Uttar Pradesh, India; Kathleen McKenna, Children's Memorial Hospital, Chicago, Illinois; Claudia Mehler-Wex, Julius-Maximilians-Universität Würzburg, Würzburg, Germany; Eberhard Meyer, Klinik für Psychiatrie und Psychotherapie des Kindes- und Jugendalters (KPPKJ) Philippshospital, Riedstadt, Germany; Dragan Mitrovic, Institute of Psychiatry, Clinical Center Novi Sad, Novi Sad, Serbia; Aida Muncada, National Center for Mental Health, Mandaluyong City, Philippines; S. Mustafa, Pacific Institute of Medical Sciences, Kirkland, Washington; Jitendra Nagpal, VIMHANS, New Delhi, India; R.J. Nichol, Free State Psychiatric Complex, Bloemfontein, South Africa; Americo F. Padilla, Miami, Florida; Anjali Pathak, Ten Broecke Hospital Research Department, Jacksonville, Florida; Yuriy Popov, Psychoneurology Institute Bekhterev, St Petersburg, Russia; Smiljka Popovic Deusic, Institute for Mental Health, Belgrade, Serbia; Fritz Poustka, J.W. Goethe University, Frankfurt, Germany; Herman Pretorius, Weskoppies Hospital, Pretoria, South Africa; Humberto Quintana, Louisiana State University Health Science Center/Division of Child & Adolescent Psychiatry, New Orleans, Louisiana; Andrzej Rajewski, Klinika Psychiatrii Dzieci I Mlodziezy AM, Poznan, Poland; Rakesh Ranjan, Rakesh Ranjan, MD & Associates, Inc., Beachwood, Ohio; Elias Sarkis, Sarkis Clinical Trials, Gainesville, Florida; Russell Scheffer, Children's Hospital of Wisconsin, Milwaukee, Wisconsin; Franco Sicuro, Millennium Psychiatric Assoc, LLC, St. Louis, Missouri; Linmarie Sikich, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina; David Spiegel, Brighton Research Group, LLC, Virginia Beach, Virginia; Brent Stevenson, Green Oaks Center for Neuropsychiatric Study, McKinney, Texas; Ahmad Sulaiman, University of Malaya Medical Centre, Petaling Jaya, Malaysia; Chin Lee Toh, KL, Hospital of Kuala Lumpur, Kuala Lumpur, Malaysia; JK Trivedi, King George Medical University GM and Associated Hospitals, Lucknow, Uttar Pradesh, India; Madeleine Valencerina, College Hospital, Cerritos, California; Petro Vlasovich Voloshin, Academy of Medical Sciences, Kharkov, Ukraine; Roger Vogelfanger, Compass Intervention Center, Memphis, Tennessee; Marianne Wamboldt, The Children's Hospital, Denver, Colorado; Johnny Williamson, Community Mental Health Council, Inc., Chicago, Illinois; Kashinath Yadalam, Lake Charles Clinical Trials, Lake Charles, Louisiana.
Appendix
| Quetiapine 400 mg/day (n=73) | Quetiapine 800 mg/day (n=74) | Placebo (n=75) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Change in BMI category | Change in BMI category | Change in BMI category | ||||||||||
| BMI category | Baseline (n) | Unchanged (n, %) | Change to lower category (n, %) | Change to higher category (n, %) | Baseline (n) | Unchanged (n, %) | Change to lower category (n, %) | Change to higher category (n, %) | Baseline (n) | Unchanged (n, %) | Change to lower category (n, %) | Change to higher category (n, %) |
| Healthy weight (5th to <85th percentile) | 51 | 50 (98.0%) | 0 | 1 (2.0%) | 44 | 43 (97.7%) | 0 | 1 (2.3%) | 46 | 43 (93.5%) | 1 (2.2%) | 2 (4.4%) |
| At-risk of overweight (85th to <95th percentile) | 7 | 7 (100%) | 0 | 0 | 12 | 9 (75.0%) | 0 | 3 (25.0%) | 13 | 12 (92.3%) | 1 (7.7%) | 0 |
| Overweight (≥95th percentile) | 10 | 10 (100%) | 0 | NA | 10 | 9 (90.0%) | 1 (10.0%) | NA | 9 | 8 (88.9%) | 1 (11.1%) | NA |
Available data presented for healthy weight, at-risk of overweight, and overweight groups.