
Abstract
In recent years, there has been an increase in pediatric clinical trials as the result of an identified need for greater research with this population. Given the potential risks, and the vulnerability of the population, there has also been an identified need for greater safety elicitation and monitoring in pediatric psychopharmacology trials, for example, through the use of a data and safety monitoring board (DSMB). However, research indicates that pediatric trials and psychiatric trials are less likely to use a DSMB. The rationale for the current study was to determine what safety methodologies have been reported in pediatric psychopharmacology trials over the past 10 years. A literature review was conducted of all pediatric psychopharmacology trials published since 2001. Results indicated that the most common elicitation method was collecting laboratory information and vital signs. Six percent of trials solely relied on spontaneous reporting of adverse events, and only 11.8% reported using a DSMB. These results suggest that elicitation methods and use of DSMBs are still low. Practical considerations, affected stakeholders, and barriers are discussed. Recommendations for moving forward include the use of multiple elicitation methods and automatic requirement of a DSMB for pediatric psychopharmacology trials, required completion of a standardized safety reporting form, and engaging multiple interested parties in these processes.
Introduction
One side of this ethical dilemma has been addressed by ensuring that more research is done in pediatric populations. The United States Food and Drug Administration (FDA) instituted the pediatric exclusivity rule which can require and incent pharmaceutical manufacturers to conduct drug trials with pediatric populations, in some cases, in exchange for 6 months of marketing exclusivity (Woodcock 2001; United States Food and Drug Administration 2009). However, the other side of this ethical dilemma, ensuring safety and protecting against potential harms associated with drugs tested in these trials, has not been adequately addressed. In order to do so, there must be adequate elicitation methods for gathering safety information, as well as monitoring methods to identify emerging trends in psychopharmacology trials. The justification for this is undeniable. An estimated 11% of pediatric clinical trials have a moderate-to-severe adverse drug reaction, ranging from required or prolonged hospitalization to permanent disability or death (Sammons et al. 2008). Justification for adequate elicitation methods also comes from the nature of the patient population. Many pediatric patients, especially those with a pervasive mental illness (e.g., autism), may have limited communication abilities and/or lack the insight required to recognize and report adverse events or significant changes in how they feel (Barlow et al. 2006). Reliance on a third party such as a parent or teacher to report changes in a child's symptoms may overlook symptoms that cannot be communicated by the child. With this vulnerable population, often limited by their developmental stage and/or cognitive ability, and the need for third-party reporting, reliance on spontaneous reporting of adverse events is not appropriate nor is it ethical. More proactive measures must be taken to elicit, rather than respond to, potential adverse events in pediatric psychopharmacology trials.
Elicitation
Adverse events may be elicited with varying levels of rigor. General inquiry methods address nonspecific side effects and may be done through open-ended questions such as “How has your child's health been since the last visit?” (Greenhill et al. 2003). General inquiry may also include the use of a rating scale or checklist for potential side effects. Drug-specific checklists, which elicit known potential side effects of the particular drug or class of drugs may also be used (Greenhill et al. 2003). Laboratory information and vital signs, such as blood pressure, height, and weight to detect changes in growth and/or significant weight gain or loss, electrocardiograms (ECGs) to detect changes in cardiac functioning, and complete blood counts to determine changes in hematology, among other measures, may also be used. Evidence to suggest if one particular elicitation method is better than another at detecting adverse events is still inconclusive (Greenhill et al. 2003).
Monitoring
Ensuring safety of research participants does not stop at elicitation. Once the information is gathered, it must be monitored in order to detect emerging trends that may indicate harm, effectiveness, or ineffectiveness. One method for doing this is through a data and safety monitoring board (DSMB). DSMBs, also commonly referred to as data monitoring committees or safety monitoring committees, among many other names, are bodies responsible for ongoing analyses of effectiveness and safety of trial drugs (Conti 1995). The DSMB can recommend that a trial be terminated early if there is sufficient evidence that the trial drug is clearly harmful, ineffective, or effective, that is, that continuing on with the trial would no longer be ethical (Conti 1995). DSMBs are typically recommended for large, multicenter trials with high morbidity and mortality rates and with vulnerable populations (National Institutes of Health 1998, 2000; United States Food and Drug Administration 2006).
The use of elicitation methods and uptake of DSMBs and interim safety analyses for safety monitoring in clinical trials has been slow and fragmented. In a previous review of pediatric psychopharmacology trials, Greenhill et al. (2003) found that 33% of trials reported using a general inquiry method (or no elicitation method whatsoever), 23% of trials reported using drug-specific checklists, and 44% reported using laboratory information and vital signs; however, reporting of safety monitoring was generally inconsistent and fragmented. A review of randomized controlled clinical trials (RCTs) published in 2000, found that only 18% of trials reported using a DSMB, 16% had planned interim safety analysis, and 24% reported at least one of the above (Sydes et al. 2004). What is particularly concerning is that RCTs in the realm of psychiatry published in general medical journals reported a DSMB in only 3% of trials versus an average of 25% across all disease categories. No psychiatric RCTs published in specialist medical journals reported a DSMB compared with 13% of RCTs across all disease areas combined (Sydes et al. 2004). These data suggest that safety in psychiatric trials is underaddressed relative to other disease areas. A review of pediatric clinical trials from 1996 to 2002 found that only 2% of trials reported a DSMB (Sammons et al. 2008). Fernandes et al. (2009) found that 13% of pediatric trials reported a DSMB in trials published between 2005 and 2007, an encouraging, yet still minor, increase from Sammons and colleagues' 2008 data. However, this may not be a true increase in DSMB use, as Fernandes and colleagues (2009) included all therapeutic interventions (e.g., behavioral interventions, procedures) whereas Sammons included pharmacological interventions only. These results, 2% and 13%, respectively, are much lower than the 18% reported in Sydes et al. (2004). Results from Fernandes et al. (2009) and Sammons et al. (2008) suggest that pediatric trials, like psychiatry trials, are less likely to take precautions against harm through using a DSMB.
The research to date suggests that despite their extreme vulnerability and need to be protected, trials involving pediatric participants and trials involving psychiatric participants fall below the already unacceptably low average for using DSMBs to monitor safety (Sydes et al. 2004; Sammons et al. 2008; Fernandes et al. 2009). The purpose of the current study is to follow up from previous research (Greenhill et al. 2003; Sammons et al. 2008; Fernandes et al. 2009), and determine what methods are used to elicit and monitor safety data in pediatric psychopharmacology trials and if there have been improvements in safety eliciting and monitoring over the past 10 years.
Methods
Literature search
A literature search was completed using psycINFO and PubMed databases with varying combinations of the following terms: youth, child, children, adolescent, pediatric/paediatric, psychopharmacotherapy, pharmacotherapy, antidepressant, anxiolytic, anti-anxiety, antipsychotic, anti-mania, antimanic, anticonvulsant, stimulant, mood stabilizer, data and safety monitoring board, data safety committee, randomized control trial, randomized clinical trial, randomized trial, medication management, mental health, psychiatry, and psychiatric. Additionally, RCTs referred to or cited in review articles that were found using the search criteria were retrieved.
Inclusion and exclusion criteria
All RCTs of pediatric psychopharmacology for the treatment of psychiatric conditions published from 2001 to the present were included for review. The last available data on safety methodology used in pediatric psychopharmacology trials were accepted for publication in 2002, and published in 2003 (Greenhill et al. 2003); 2001 was chosen as a starting point so as not to be redundant with Greenhill and colleagues, but to ensure that there were no gaps in data reported following up from Greenhill et al. (2003). Studies that examined natural products (e.g., gingko biloba, omega-three fatty acids), for the treatment of pediatric mental illness were also included. Head-to-head trials, which compared two or more psychopharmacotherapies without the inclusion of a placebo or treatment as usual control group were also included. Trials were excluded from the analysis if any participants were >19 years of age, if randomization was not used, and if psychopharamacotherapy was used for nonpsychiatric purposes, such as for the treatment of nausea or migraine.
Data synthesis and analysis
Abstracts of all articles returned from the literature search from 2001 onwards were reviewed to determine if they met the inclusion criteria. If it was not immediately apparent from the abstract whether the study met the criteria, the methods sections were further analyzed. The last paragraph of the Introduction, Methods, and Results sections of all studies meeting inclusion criteria were reviewed for safety information. When possible, portable document formats (pdfs) of studies were searched for the terms: safety, tolerability, adverse event, side-effect, and data and safety monitoring board, in order to identify safety information. The characteristics of the study were recorded as well as the safety methodology used. Specifically, adverse event elicitation methods were recorded and coded as either: General inquiry, general inquiry using a rating scale or checklist, drug-specific checklist, or laboratory and/or vital information. Use of a data and safety monitoring board (DSMB) was also recorded, whether an interim safety analysis was performed, if there were predefined criteria for stopping a trial either because of safety or efficacy data, or if there were statistical methods for monitoring safety reported. Studies were recorded as relying solely on spontaneous reporting of adverse events if doing so was explicitly reported in the Methods section, if no safety methodology was reported in the Methods section (but adverse event information was reported in the Results section), or if no safety information was reported at all. Descriptive characteristics of each study were also recorded, which included sample size, number of sites included in the study, duration of the trial, number of treatment arms, and which treatment(s) and/or control methods were used.
Data were recorded and analyzed using Excel. Frequencies and percentages of each adverse event elicitation method, the use of a DSMB, interim safety analysis, predefined criteria for stopping the trial, and statistical monitoring methods for safety information, as well as sole reliance of spontaneous reporting, were calculated. The number of different types of safety methods used in each study was also recorded as well as the average number of different safety methodologies used overall.
Results
A total of 119 studies met inclusion criteria and were included in the analysis (see Table 1 and Appendix). The average sample size was 111 participants, and of those reporting the number of trial sites, 50% were multicentered. The average length of treatment for trials was 10.4 weeks (see Table 2).
DSMB, data and safety monitoring board.
The most common adverse event elicitation method was through laboratory and vital sign measures (75.6%), followed by drug-specific checklist (39.5%), general inquiry (35.3%), and general inquiry with checklist or rating scale (34.5%). Just <6% of studies reported that they did not proactively elicit safety information and relied solely on the use of spontaneous reporting. A total of 14 of the 119 studies (11.8%) reported using a DSMB and 9 (7.6%) reported using an interim safety analysis (8 of which also used a DSMB and 1 of which did not). Three (2.5%) reported using statistical monitoring methods for safety, and only one (0.8%) study reported having predefined criteria for terminating the trial (see Fig. 1). The percentages will not add up to 100% as the studies could use more than one method. The number of different types of safety methodologies used in studies ranged from zero (sole reliance on spontaneous reporting) to seven of the possible eight elicitation and monitoring tools recorded, with an average of two different safety methodologies used.
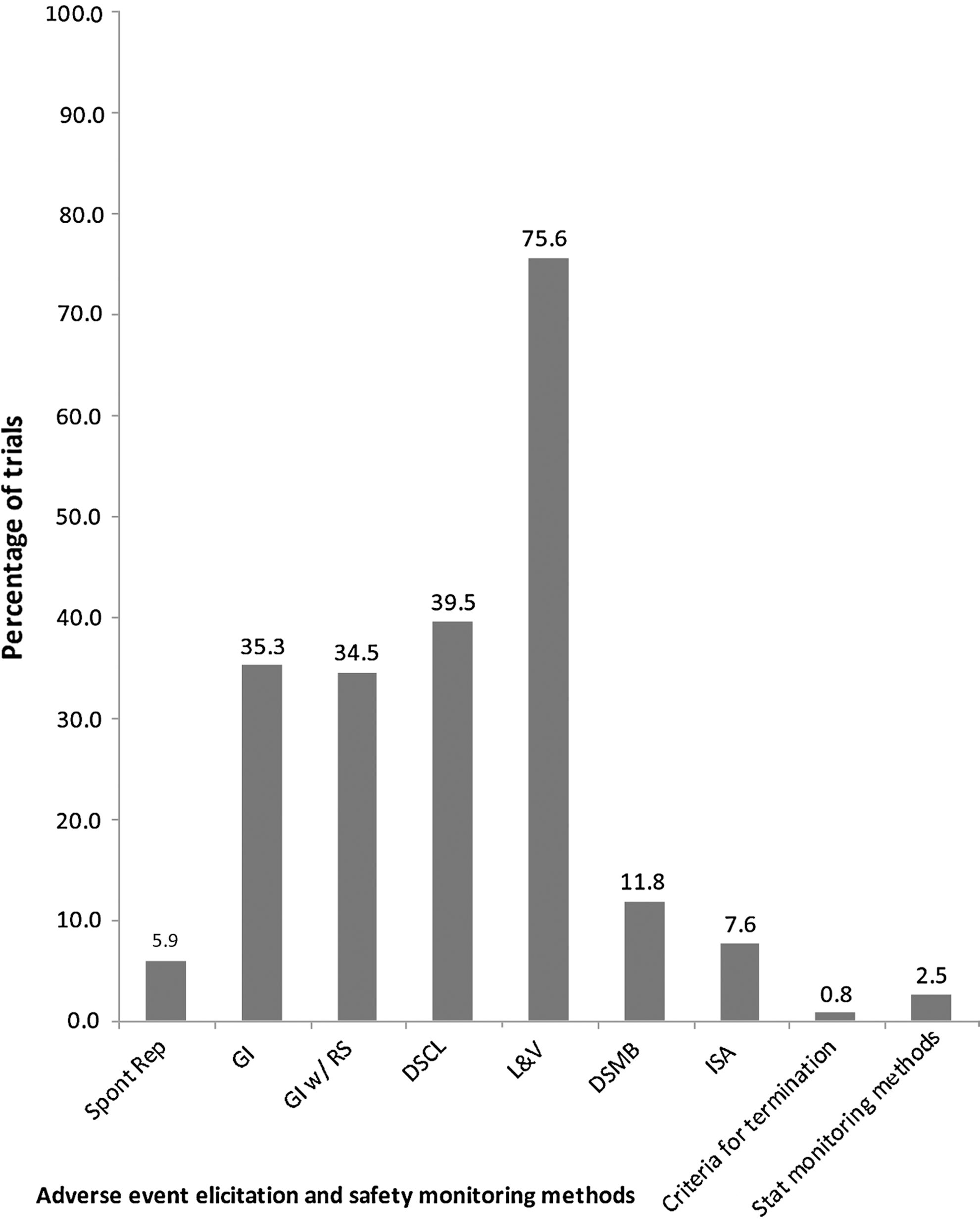
Safety elicitation and monitoring methods used in pediatric psychopharmacology trials since 2001. Spont Rep, spontaneous reporting; GI, general inquiry; GI w/ RS, general inquiry with rating scale; DSCL, drug specific checklist; L&V, laboratory and vital signs; DSMB, data and safety monitoring board; ISA, interim safety analysis; Stat, statistical.
Discussion
The results indicate that there has been an increase in the reported use of elicitation methods since 2002 (Greenhill et al. 2003) (see Fig. 2). However, the reported use of a DSMB appears unchanged. Reported DSMB use in the data was slightly less than reported by Fernandes et al. (2009) and still less than the 18% reported in Sydes et al. (2004). This indicates that pediatric psychopharmacology trials are still reporting disproportionately less DSMB use despite the vulnerability of their participant population. The reported use of an interim safety analysis was also less than generally reported (16%) in Sydes et al. (2004). Particularly alarming is that <1% reported predetermined termination criteria. This suggests that there is a lack of planning and forethought as to what constitutes enough harm, effectiveness, or ineffectiveness that it would be unethical to continue.
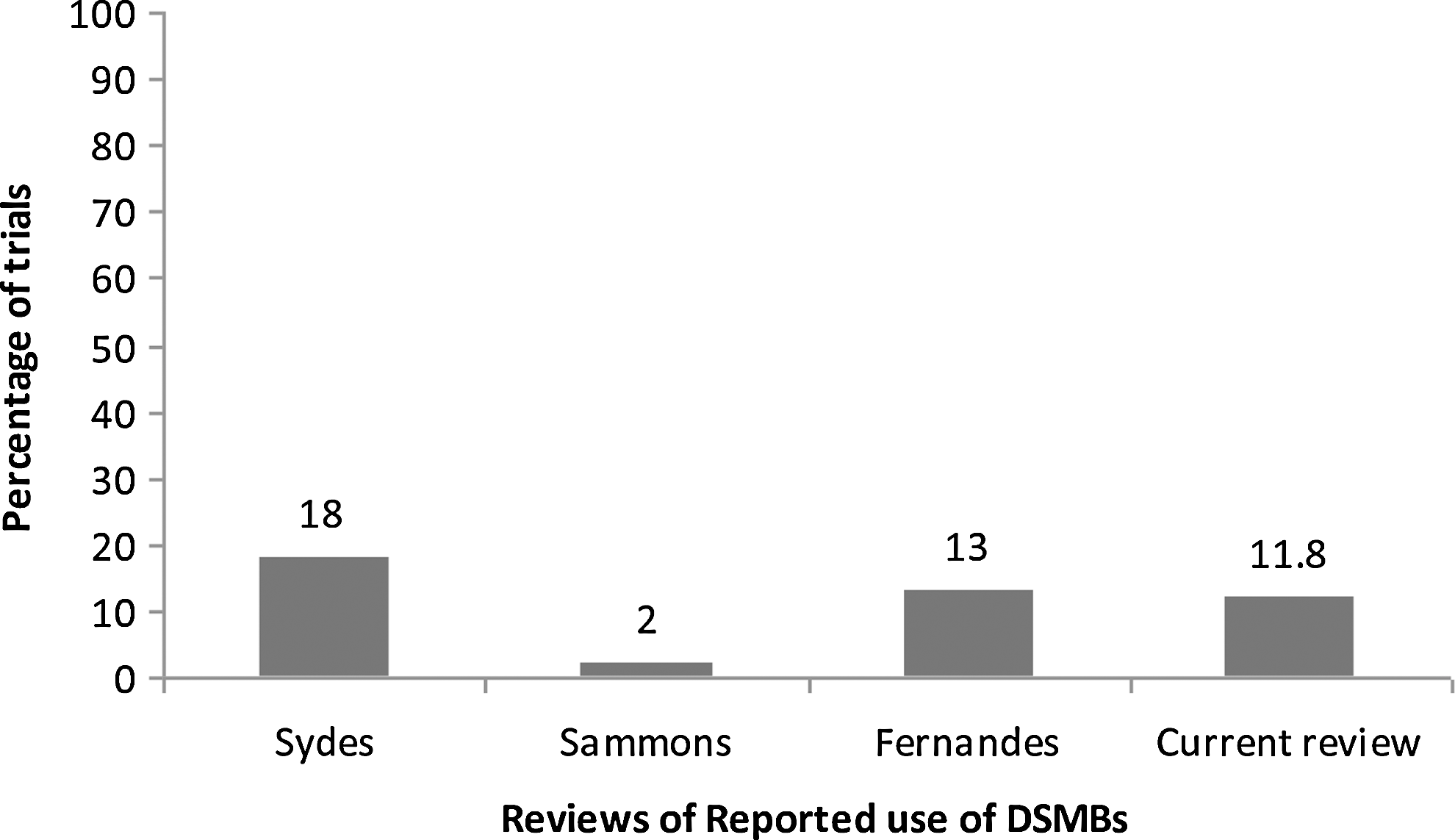
Reported use of data and safety monitoring boards from four reviews of randomized controlled trials.
The results also indicate that trials reported using, on average, two different elicitation and/or monitoring methods. Given that there is a lack of research to discriminate the relative effectiveness of elicitation methods (Greenhill et al. 2003), investigators should err on the side of caution and use several different methods. Finally, 6% of trials reported they relied solely on spontaneous reporting to gather adverse event information; this should not be happening with this population under any circumstances.
Affected stakeholders
There are many different stakeholders affected by the use, or lack thereof, of safety elicitation and monitoring methods. Buy-in and action from these stakeholders can help advance the enhancement of more rigorous safety methodologies and reporting in pediatric psychopharmacology trials. For some, enhancement of safety elicitation and monitoring will require more resources, which may act as a deterrent to doing so. However, more rigorous safety methodology can not only better protect research participants, but also lend greater credibility to the research and the product, creating a mutually beneficial situation for all stakeholders. The affected stakeholders and their perspectives are listed here.
Current and future participants, patients and parents
This is the population that stands to benefit or lose from the research that occurs. Knowing that a trial has proactive monitoring methods with appropriate expertise can increase parents' assurance of their children's safety and increase likelihood of participation (van den Ouweland et al. 2004). Furthermore, using a DSMB means that consideration for the potential harms and benefits to the patient population in general are proactively being monitored with the appropriate expertise.
Investigators and research teams
Increasing the demand and standards for safety monitoring places an extra burden on the investigators and research team. This may mean more time administering checklists to elicit adverse events, which may require training for research staff, or preparation of data to be sent to the DSMB. However, having a DSMB in place also benefits investigators by providing guidance and recommendations to them and overall enhancing the scientific credibility of their work.
Institutions
Institutions can benefit greatly from the use of a DSMB. Institutional review boards (IRBs) can require proposed studies to use a DSMB, and can alleviate some of the burden of their work. Also, DSMBs can be a source of important information to the IRB about the safety of trials occurring in their facilities, and use of a separate DSMB can result in more efficient use of skills (Califf et al. 2003). Academic health centers in particular, are affected by the use or lack of use of DSMB; with research as an integral part of their vision and mission, academic health centers are in a position of influence on the topic and can help raise the bar for safety by requiring DSMBs for appropriate trials that occur within their walls (Califf et al. 2003).
Trial sponsors and granting agencies
Sponsors are a key potential source to drive the use of DSMBs and increase the rigor of safety methodology. Other funding agencies can follow in the footsteps of the National Institutes of Health (NIH) and create regulations regarding DSMBs for trials that they fund. However, this often requires sponsors to absorb the costs of the DSMB. At an additional 5% per trial (van den Ouweland et al. 2004), this can dramatically increase the overall costs to funding agencies.
Pharmaceutical manufacturers
If a pharmaceutical company is sponsoring the trial, the added costs of a DSMB may discourage its use; however, having one can also lend greater legitimacy to the company's product. The potential affect that use of DSMBs may have on drug prices should be also considered. If pharmaceutical companies spend more to ensure that DSMBs are in place for their trials, these increased costs will likely be recovered through higher drug prices to the consumer (patient or third-party payer). Furthermore, under the FDA pediatric rule, extended marketing exclusivity for these companies who conduct trials with pediatric populations may also increase overall drug costs through decreased opportunity for generics to enter the market.
Regulators/decision makers
At the highest level of decision making, regulators such as Health Canada can take a stance and create guidelines for the use of a DSMB, or go even further and mandate its use in appropriate trials. Furthermore they can also create guidelines and/or regulations of minimum standards for elicitation methods and reporting methods. Finally, the safety methodology used in clinical trials can inform and be considered when determining if a particular drug should be on a provincial formulary, thereby encouraging more rigorous safety methodology in clinical trials.
Barriers to enhancing safety methodology in pediatric psychopharmacology trials
Despite the identified need for enhancing safety elicitation and monitoring in clinical trials via DSMBs (Greenhill et al. 2003; Carandang et al. 2007), there appears to be a gap between the identified need and the actual implementation of these recommendations. This is most likely because of a number of barriers that are listed here.
Independence and objectivity
Given the critical decisions that the DSMB must make, and the possible implications of those decisions affecting the current research participants and the patient population as a whole, they must remain objective, hence the emphasis on independence of a DSMB (Ellenberg 2001). However, to find someone with the appropriate level of expertise to participate in the DSMB, but not closely tied to the trial itself and the researchers conducting the trial, would be challenging. First of all, having expertise in the area that these people would have expertise in, also presumably means that they would have a strong opinion and potential interest in the outcome of the trial. For example, DSMB members who have expertise in the area may prefer one outcome over another in order to corroborate their own research. Second, given that in many research areas there tends to be relatively small core group of researchers and as such, the DSMB members would presumably know the investigators fairly well from conferences, and have cited their research, which would compromise the ability of those DSMB members to remain objective.
Real-world safety and effectiveness
There has been increasing focus on safety and effectiveness of drugs, once they have been tested and approved and are on the market. This is evident in Canada's National Pharmaceutical Strategy and from the formation of the Drug Safety and Effectiveness Network, which focuses solely on real-world settings (Federal /Provincial/Territorial Ministerial Task Force 2006; Canadian Institutes of Health Research 2009). With shifting attention to real-world settings, there may be fewer resources dedicated to controlled trials. However, having rigorous elicitation and monitoring methods may mean that a broader scope of participants can be recruited. For example, more participants with various comorbidities may be recruited into a study because the proper safety infrastructure and processes are in place. A caveat to this, however, is that recruiting more heterogeneous samples will compromise the scientific validity of trials which is also under the oversight of the DSMB.
Discouraging research
An unintended consequence of imposing higher safety standards for clinical trials may be discouraging researchers from conducting these trials. As previously discussed, this would be unethical and would deprive the pediatric population of the opportunity to benefit from research (Committee on Clinical Research Involving Children 2004). However, enhancing safety methodology, especially using a DSMB, can also be an opportunity for researchers, lending more legitimacy and credibility to their findings.
Difficulty assessing safety compared with assessing effectiveness
Despite many controlled clinical trials claiming to evaluate both the effectiveness and safety of therapeutic interventions, noticeably less attention is given to safety and how adverse event information is elicited, how it is monitored and analyzed, and the implications. This may be because of the increased difficulty and complexity in evaluating safety, which is often exploratory and must sometimes answer unexpected questions; a stark contrast to effectiveness, which is explored through predetermined methods to answer one specific research question (Greenhill et al. 2003).
Resources
Implementing DSMBs will obviously require time and human and financial resources; a particular challenge with rising costs and limited public funds. More rigorous elicitation requires extra time and appropriately trained researchers to administer questionnaires and enter data. A DSMB requires expertise and remuneration for its work and time. However, the costs of not investing in safety in the early stages, before a drug is on the market, must also be considered. The potential costs associated with adverse drug reactions such as hospitalization, disability, and death are presumably significant, especially with a pediatric population, where there would be accumulating healthcare costs, lost productivity, and potential life years lost.
Recommendations for moving forward
After reviewing the affected stakeholders and potential barriers to enhancing safety methodology in pediatric psychopharmacology trials, we propose several recommendations for moving forward.
Multiple adverse event elicitation methods
Sole use of spontaneous reporting should never be permitted with pediatric psychopharmacology trials. This can be enforced at multiple levels, including IRBs, sponsors, and regulators. Multiple elicitation methods should be used, including a general inquiry question to account for anything that may be missed by a checklist. Drug-specific checklists should be used where they exist; if one does not exist, then general checklist should be used. Laboratory information and vital signs should also be collected to address physical side effects. Although spontaneous reporting should never be the only source of safety information, there should be methods and processes in place that easily allow participants and parents to spontaneously report adverse events. For example, having the ability to contact someone on the research team 24 hours a day, 7 days a week to report adverse events, or having advanced access to healthcare appointments for those participating in high-risk research can facilitate reporting of side effects. Although use of all of these elicitation methods may seem extreme or potentially inefficient, research is not yet clear about which method is most sensitive (Greenhill et al. 2003). Until there is evidence to support one particular method over another, investigators should err on the side of caution and use all methods at their disposal to ensure thoroughness in their safety assessment.
Automatic requirement of DSMBs for pediatric psychopharmacology trials
Given the vulnerability of the population, all RCTs of psychopharmacology in a pediatric population should automatically be required to have a DSMB (Sammons et al. 2008). This has already been required for NIH-funded trials and recommended by the FDA (National Institutes of Health 1998, 2000; United States Food and Drug Administration 2006). Canadian regulatory bodies (e.g., Health Canada) and funding agencies (e.g., Canadian Institutes of Health Research [CIHR]), should follow suit and create more rigorous criteria for running and receiving funding for clinical trials investigating pediatric psychopharmacology. The DSMB for multisite trials should be centralized to avoid duplication of efforts with the IRBs and clear terms of reference with reporting relationships and liability should be outlined prior to the beginning of the trial.
Standard safety reporting form
Safety methodology in clinical trials should be clear and transparent. This can be a challenge for reporting safety methodology given the limited space for publications. A standardized safety reporting form should be developed that is required for all investigators to complete when submitting a manuscript for publication and available to everyone reading the publication. This should include, at a minimum, which elicitation methods were used, how often they were used, the particular questions used for general inquiry, whether a DSMB was used, how many interim safety analyses were performed, and any changes or recommendations from the DSMB. Increased transparency of safety methodology should encourage the use of enhancement of safety methods. Furthermore, having this information reported in a standardized format that is user friendly can reduce the burden on investigators reporting the information, facilitate research on which elicitation methods are the most effective, and reduce the fragmented and elusive reporting as it currently stands.
Engage multiple stakeholders
There are already many bodies with an interest in this area (e.g., Canadian Council for Protection of Human Research Participants, The National Council on Ethics in Human Research, Canadian Patient Safety Institute). These groups should work together along with investigators, parents, participants, funding agencies (e.g., CIHR) and regulators (Health Canada) to advance the agenda for enhancing safety methodology in clinical trials with vulnerable populations. Given these interested parties have may have varying levels of support for the enhancement of safety methodology in pediatric psychopharmacology trials, appropriate incentives and regulations should be considered.
Training/education of DSMB members
Formal education of DSMB members is important, because of the responsibility of the DSMB for the safety of study participants. DSMB members should be trained in a standardized manner so that safety methodology can be applied across studies and replicated. From this, safety methods and analysis that work can be further developed, and those that do not work can be eliminated. Standardized training would benefit the field by ensuring that all studies have the benefit of the latest safety methodology deduced from experience and research. Currently, many DSMB members do not get formal training, and rely too heavily on the investigators they are supposed to monitor. Detailed information on DSMBs can be read in detail elsewhere (Ellenberg et al. 2002; Slutzsky and Lavery 2004). Also, the FDA has a pdf on DSMBs:
Limitations
There are several limitations of the study that must be noted. First of all, the literature search, although thorough, was not exhaustive. Only two search engines were used. A more thorough search would use more databases in addition to hand-searching relevant journals. However, even if this was not an exhaustive search, it provides a robust sample of pediatric psychopharmacology trials over the past 10 years. Also, a wider array of search terms could have been used, especially given the vast nomenclature for DSMBs; however, research suggests that “DSMB” is the most commonly used name (Fernandes et al. 2009). Furthermore, the intent was to find all randomized control trials, not just those that used a DSMB; therefore, this impact should be considered minimal. Studies not published in English were also excluded.
All data collection and coding was done by one researcher and no inter-rater reliability was calculated. Although this is a limitation, agreed-upon operational definitions were decided by the research team, which should reduce the possibility of errors in the data collection and coding.
Another limitation is that these results are based on only what was reported in published trials. No authors were contacted to ask about their safety methodology. A future study may consider choosing a small random sample of studies among those that did not report using a DSMB, and attempt to contact the investigators to see if a DSMB was actually used. Presumably, one hopes, the results from this study are an underestimation of the safety monitoring in clinical trials. However, this reiterates Greenhill et al.'s (2003) findings and conclusion that there needs to be more standardized reporting of safety methodology. This is especially relevant given that many clinical trials state that their purpose is to assess safety in addition to efficacy, and some even incorporate “safety” or “tolerability” into their title, despite reporting limited information on the topic. Also, many of the trials were ambiguous in their descriptions of some methodologies; for example, some trials stated that “adverse events were recorded,” which does not give insight into how or if they were elicited. In these instances, trials were coded as relying solely on spontaneous reporting even though they may have actively elicited the information. However, in vague descriptions such as the above, the investigators decided to err on the side of caution and assume that if something was not reported, it was not done. This ambiguity, again, lends further support to having standardized reporting mechanisms. Also, only published trials were included, and as such, the results cannot be generalized to unpublished trials, and results may be skewed positively or negatively as a result.
A final limitation may be that our results are an underestimation. When asked about their most recent experience taking part in a DSMB for a clinical trial, 62% of researchers and biostatisticians' reported that they had predefined criteria for termination (Tereskerz et al. 2011). In addition, the number of DSMBs actually utilized is most likely underestimated, as this may not be overtly stated in the outcome papers. These omissions of DSMB utilization may be the result of factors such as journal editors' not requiring this information in manuscripts, or only reporting use of a DSMB when the population being studied is deemed high risk.
Conclusions
Research in pediatric psychopharmacology is ethically necessary, and this vulnerable population is owed a high level of safety during their participation. Despite the identified need (Greenhill et al. 2003; Carandang et al. 2007), the current study suggests that little progress in the way of safety monitoring and safety reporting has been made. This may be attributable to the numerous affected stakeholders, the required resources, difficulty finding the appropriate level of expertise and independence, and a shifting focus on real-world safety and effectiveness.
Despite the seemingly stagnant progress in this area, there have been more recent discussion, guidelines, regulations, and publications from regulators, sponsors, and researchers on how safety should be addressed and reported and how DSMBs should operate. This increasing attention, along with the current recommendations, will, we hope, provide a foundation for moving forward and enhancing safety in pediatric psychopharmacology trials.
Clinical Significance
Focusing on safety methodology in research studies would help clinicians determine the safety risks posed by psychotropic medications in the pediatric psychiatry population. The unknown safety profile of many psychotropics in the pediatric psychiatry population potentially exposes children and adolescents who take these psychotropics to unknown safety risks. Many of the studies analyzed here are only efficacy studies, ignoring safety risks; therefore, how can these studies be applied clinically if the safety risks are unknown?
Footnotes
Acknowledgments
We thank Joyce Totton, Darlene Boliver, and Karen Comeau for their input in the development of this study.
Disclosures
For the past 5 years, Dr. Carandang has served on the Advisory Board of Shire, received an educational grant from Pfizer, and received Honoraria from Purdue, Bristol-Myers Squibb, and Janssen. Ms. Yuill has no conflicts to disclose.