
Abstract
Objective:
The most widely utilized pharmacological treatment of attention-deficit/hyperactivity disorder (ADHD) is the psychostimulant methylphenidate (MPH). Most MPH formulations consist of the racemic mixture of d-threo-(R, R)-MPH and l-threo-(S, S)-MPH isomers. MPH is characterized by its low bioavailability and short half-life (2–3 hours). Additionally, significant inter-individual variability in MPH pharmacokinetics has been consistently documented. Accordingly, efforts have been directed at developing alternatives to MPH as therapeutic agents. A wide range of MPH analogues (dl-α-[2-piperidyl]-phenylacetic acid esters) have been synthesized with the dopamine transporter (DAT) and norepinephrine transporter (NET) as principle neuropharmacological targets. The present study investigated the metabolic profiles and pharmacological activity of the isopropyl ester derivative of MPH, dl-isopropylphenidate (IPH), both in vitro and in vivo.
Methods:
The synthesis, monoaminergic transporter binding, cellular uptake profiles, and assessment of metabolic hydrolysis and transesterification in the presence of ethanol are described using MPH as a comparator. Additionally, an in vivo assessment of IPH stimulant effects (vs. saline) in rats was performed with locomotor activity as a pharmacodynamic outcome.
Results:
IPH displayed unique pharmacological characteristics including greater DAT than NET binding and cellular uptake activity, and greater resistance to hydrolysis and transesterification via carboxylesterase 1 relative to MPH. Further, sustained psychostimulant properties offer the prospect of an enhanced duration of action.
Conclusions:
Our findings are consistent with IPH exhibiting attributes distinguishing it from MPH and warranting further study and development of IPH as a novel psychotherapeutic agent.
Introduction
A
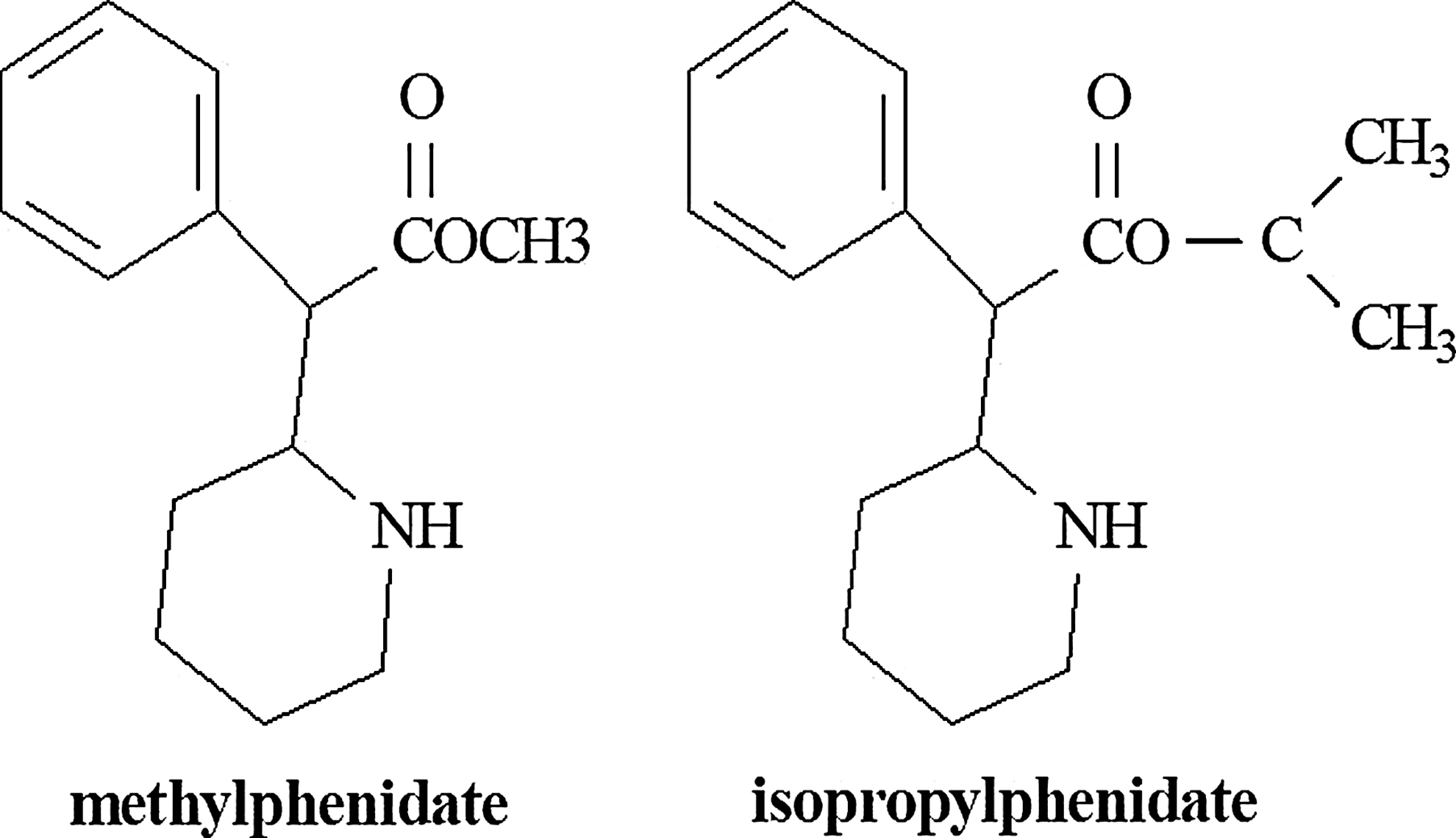
Chemical structures of methylphenidate (MPH) and isopropylphenidate (IPH).
In the present study, the MPH homolog dl-isopropylphenidate (IPH) (Fig. 1) was synthesized and assessed for its pharmacological activity and metabolic profiles. Monoamine transporter binding and cellular uptake experiments were performed on racemic IPH, as well as on the comparators MPH and the ethyl ester congener previously characterized by our group, dl-ethylphenidate (EPH) (Patrick et al. 2005b). Additionally, an assessment of metabolic hydrolysis of the three compounds by CES1 was performed utilizing CES1 transfected cell lines, human liver microsomes (HLM), and human intestinal microsomes (HIM). Furthermore, the potential for a CES1-mediated transesterification reaction between IPH and ethanol to ethylphenidate was evaluated and compared with that of MPH in vitro, in view of the metabolic drug interaction associated with the MPH–ethanol combination (Patrick et al. 2007). Finally, an in vivo assessment of IPH stimulant effects (vs. saline) on locomotor activity was performed utilizing experimental methodology previously employed to establish the in vivo locomotor activity of the separate d- and l-isomers of MPH in rats (Patrick et al. 1987). The overarching goal of this series of experiments was to explore the pharmacological characteristics of IPH as a potential new therapeutic entity.
Materials and Methods
Chemical compounds and animals
MPH was purchased from Sigma-Aldrich (St. Louis, MO). EPH synthesis was conducted in our laboratory as previously described (Patrick et al. 2005b). IPH was synthesized in our laboratory using the following method: Briefly, (±)-ritalinic acid (2 mM) was dissolved in isopropyl alcohol saturated with HCl gas (75 mL) and refluxed for 24 hours under nitrogen. The solution was evaporated to dryness under reduced pressure and purged with nitrogen, and the white residue was then dissolved in a minimum volume of warm isopropyl alcohol. Diethyl ether was then added to turbidity and the flask was stored for 24 hours at 2°C. The resulting white crystalline product was filtered, washed with diethyl ether, and dried under vacuum. The purity of the synthetic material was confirmed by gas chromatography-mass spectrometry. All compounds studied were assessed as their HCl salts. The remaining reagents and solvents were of the highest grade commercially available. Animals used in these experiments were male Sprague–Dawley rats weighing 200–300 g and obtained from Charles River Laboratories (Wilmington, MA).
Transporter binding and cellular uptake
All described monoamine transporter binding studies as well as cellular uptake assays were performed in duplicate by CEREP (Celle l'Evescault, France) and are described subsequently. Further details of all assays performed may be accessed at the CEREP web site (
Monoamine transporter radioligand binding assays
Dopamine transporter radioligand binding assay
Evaluation of the affinity of all three compounds for the human DA transporter (DAT) in transfected Chinese hamster ovary (CHO) cells was determined in a validated radioligand binding assay. Cell membrane homogenates were incubated for 120 minutes at 4°C with 0.5 nM [3H] GBR 12935 in the absence or presence of each test compound in a standard buffer solution. Nonspecific binding was determined in the presence of 10 μM N-(1-[Benzo[b]thien-2-yl-cyclohexyl]) piperidine (BTCP). Following incubation, the samples were filtered under vacuum through glass filters and rinsed several times with ice-cold 50 mM Tris-HCl using a 96 sample cell harvester. The filters were then dried and measured for radioactivity with a scintillation counter (TopCount, Packard) using a liquid scintillation cocktail (Microscint 0, Packard). The results of DAT binding experiments as well as the other transporter assays described in the following sections were expressed as a percent inhibition of the control radioligand specific binding. The standard reference compound was BTCP, which was tested in each experiment at several concentrations in order to obtain a competition curve from which its inhibitory concentration (IC)50 was calculated.
Norepinephrine transporter radioligand binding assay
Evaluation of the affinity of compounds for the human NE transporter (NET) in transfected CHO cells was determined in a radioligand assay analogous to the procedures described for the DAT assay. Cell membrane homogenates were incubated for 90 minutes at 4°C with 1 nM [3H] nisoxetine in the absence or presence of each test compound in a standard buffer solution. Nonspecific binding was determined in the presence of 1 μM of desipramine. Following incubation, the samples were filtered rapidly under vacuum and rinsed several times with a buffer solution. The filters were then dried, and radioactivity counts were obtained. The results are expressed as percent inhibition of the control radioligand specific binding. The standard reference compound was the tricyclic antidepressant (TCA) protriptyline, which was tested in each experiment at several concentrations to generate a competition curve from which its IC50 was calculated.
Serotonin transporter radioligand binding assay
An evaluation of the respective compounds was also conducted regarding their affinity for the human serotonin 5-HT transporter (SERT) in transfected CHO cells. Briefly, cell membrane homogenates were incubated for 90 minutes at 4°C with 2 nM of [3H] imipramine in the absence and presence of each of the assessed compounds. Nonspecific binding was determined in the presence of 10 μM of imipramine. After incubation, the samples were filtered rapidly under vacuum and rinsed several times with buffer solution. The filters were then dried and measured for radioactivity via scintillation counter. The standard reference compound was the TCA imipramine, which was tested in each experiment at several concentrations to generate a competition curve from which its IC50 was calculated.
Cellular-based monoamine uptake assays
Norepinephrine uptake
The evaluation of the effects of each compound of interest (MPH, EPH, IPH) on NE uptake utilized synaptosomes prepared from the rat hypothalamus. These synaptosomes (100 μg) were incubated for 20 minutes at 37°C with 0.1 μCi [3H]norepinephrine in the absence (i.e. control) or presence of the test compound or the reference compound in a standard buffer solution. Basal control activity was determined by incubating the same mixture for 20 minutes at 0°C in the presence of 10 μM protriptyline to block the uptake. Following incubation, the samples were filtered, counted using a scintillation instrument, and the results expressed as a percent inhibition of the control uptake of [3H]norepinephrine. The standard inhibitory reference compound was the TCA protriptyline, which was tested in each experiment at several concentrations to obtain an inhibition curve from which its IC50 value was calculated.
DA uptake
The evaluation of the effects of the three compounds on DA uptake again utilized synaptosomes, but this time prepared from the rat striatum. The synaptosomes were incubated for 15 minutes at 37°C with 0.1 μCi [3H]dopamine in the absence and presence of the test compound or the reference compound in a buffer standard buffer solution. Basal control activity was determined by incubating the same mixture for 15 minutes at 4°C in the presence of 1 μM GBR12909 to block the uptake. After incubation, the samples were filtered, counted, and the results expressed as a percent inhibition of the control uptake of [3H]dopamine by scintillation count. The standard inhibitory reference compound was GBR12909, which was tested in each experiment at several concentrations to obtain an inhibition curve from which its IC50 value was calculated.
Serotonin uptake
The assessment of the effects of the three compounds on 5-HT uptake utilized measures of [3H]5-HT incorporation into synaptosomes prepared from the rat brain. The synaptosomes were incubated for 15 minutes at 37°C with [3H]5-HT (0.2 μCi/mL) in the absence and presence of each of the three assessed compounds or the reference compounds. Following incubation, the samples were filtered, counted using a scintillation instrument, and the results expressed as a percent inhibition of the control uptake of [3H]5-HT. The standard inhibitory reference compound was the TCA imipramine, which was tested in each experiment at several concentrations to obtain an inhibition curve from which its IC50 value was calculated.
Determination of relative hydrolytic rates of MPH, EPH, and IPH
The assessment of relative rates of hydrolysis of the three ester homologs utilized CES1 transfected cell lines, HLM, and HIM. The s9 fractions from the HEK293 cells transfected with human CES1A1 gene were prepared utilizing a method developed in our laboratory and described previously (Zhu et al. 2008). Hydrolysis of IPH, EPH, and MPH was assessed by measuring the formation of the hydrolytic metabolite RA following incubation with cell s9 fractions, HLM, and HIM. Briefly, 50 μL of freshly prepared substrates (racemc IPH, EPH, and MPH) was mixed with 50 μL of enzymes in 1.5 mL Eppendorf tubes. Both substrates and enzymes were diluted in Dulbecco's Phosphate-Buffered Saline (DPBS, 20 mM HEPES, pH 7.4). The final substrate concentration was 1 mM, and the final enzyme concentrations were 0.5 mg/mL, 0.2 mg/mL, and 0.2 mg/mL for the s9 fractions, HLM, and HIM, respectively. Following incubation at 37°C for 60 minutes, the reaction was terminated by adding 500 μL of methanol. The samples were then centrifuged at 16,000g at 4°C for 5 minutes to remove precipitated protein. The concentrations of the common hydrolytic metabolite RA of all three compounds were determined utilizing a validated high-performance liquid chromatography (HPLC) method we previously described (Zhu et al. 2008).
CES1 catalyzed transesterification IPH and MPH in the presence of ethanol
The working solutions of the s9 fractions of CES1 transfected cells and the substrates (MPH, IPH, and ethanol) were prepared in DPBS containing 20 mM HEPES (pH 7.4). The reaction was initiated by mixing 200 μL of the s9 fractions, 100 μL of MPH or IPH, and 100 μL of ethanol. The final concentrations of the s9 fraction protein, MPH, IPH, and ethanol were 2 mg/mL, 1mM, 1mM, and 10 mM, respectively. After incubation at 37°C for 1h, the reaction was terminated by adding a fourfold volume of ice-cold methanol. The samples were then briefly vortexed and centrifuged at 16,000g for 5 minutes at 4°C, and the resulting supernatant was subjected to HPLC analysis to assess the concentrations of the transesterification product EPH from both MPH and IPH, and common metabolite RA. The s9 fractions prepared from the vector transfected cells were included as a negative control for any nonenzymatic hydrolysis that might occur. The CES1-mediated metabolism of MPH and IPH (hydrolysis and transesterification) were estimated by substracting the amounts of RA and EPH produced in the vector s9 samples from that observed in the CES1 s9 fraction samples.
Locomotor activity measurement in rats
Locomotor-inducing activity of IPH was measured according to methods previously described for the differential pharmacology of MPH enantiomers (Patrick et al. 1987). The present study compared only the IPH homolog versus saline dosing since the other compounds have been extensively investigated in vivo previously (Williard et al. 2007). In brief, following an initial 60 minute habituation period to the activity chamber, racemic IPH (or saline) was administered intraperitonealy (i.p.) to each animal (n=5 per active drug group; n=3 per saline group) at a dose of 10 mg/kg to correspond with 10 mg of racemic MPH (delivering 5 mg of d-MPH), which was previously determined to produce near maximal behavioral responses (Patrick et al. 1987). Locomotor activity was recorded within doughnut-shaped cages with six photocell sensors equally spaced around a 9 cm runway. Activity counts as assessed by light beam interruptions were recorded for each animal in increments of 10 minutes over a 2 hour period. Differences in the cumulative motor activity accounts were assessed by the unpaired, two tailed Student t test. The level of significance was set at p<0.05.
Results
Monoamine transporter binding and cellular uptake studies
The binding of racemic IPH, MPH, and EPH to the prominent cellular monoamine transporters DAT, NET, and SERT revealed that, as a group, binding affinities were greatest for DAT. All compounds produced significant effects at the DAT, with insignificant differences noted among the three compounds (Table 1). With regard to NET, as anticipated, MPH exhibited substantial binding as measured by inhibition of specific control binding, whereas EPH exhibited approximately half the binding affinity of MPH, and IPH was the lowest at a value approximately one third that of MPH at the tested concentration (10 μM). In the case of SERT, none of the compounds assessed either approached or exceeded 50% inhibition of control specific binding.
BTCP, N-[1-(Benzo[b]thien-2-yl-cyclohexyl)]piperidine; [3H]GBR-1293, 1-[2-(Diphenylmethoxy)ethyl]-4-(3-phenylpropyl)piperazine; MPH, racemic (dl) methylphenidate; EPH, racemic ethylphenidate; IPH, racemic isopropylphenidate.
The results of monoamine cellular (i.e., functional) assays are presented in Table 2. With regard to DA, all three compounds exhibited significant effects on the uptake of this monoamine, with little difference observed among the agents. Norepinephrine uptake studies indicated that MPH and EPH exerted the most effects, whereas IPH was significantly lower at the concentration of 10 μM. Finally, 5-HT uptake was not affected to any significant degree by any of the three assessed compounds.
MPH, racemic (dl) methylphenidate; EPH, racemic ethylphenidate; IPH, racemic isopropylphenidate.
Hydrolysis of IPH, EPH, and MPH
CES1 is highly expressed in the human liver, and is purported to account for the majority of hydrolytic activity within the organ, whereas CES2 is the predominant hydrolase in the human intestine (Kagan and Hoffman 2008). In the present study, the susceptibility of MPH, EPH, and IPH to CES1-mediated hydrolysis was investigated in parallel by incubation of each substrate with the cell s9 fractions of CES1 transfected cells as well as HLM and HIM preparations. The results indicated that the catalytic efficiency of CES1-mediated hydrolysis upon MPH is approximately 10-fold higher than for IPH (Fig. 2). When HLM preparations were utilized for incubations, EPH appeared to be the most vulnerable substrate relative to MPH and IPH, whereas IPH was the most resistant substrate to HLM (Fig. 2). Data from the HIM incubation studies where CES2 (but not CES1) was extensively expressed indicated that EPH was an efficient substrate of CES2. In contrast, catalytic activity of HIM was extremely low with regard to both MPH and IPH hydrolysis (Fig. 2).

Hydrolysis of methylphenidate (MPH), ethylphenidate (EPH), and isopropylphenidate (IPH) by carboxylesterase 1 (CES1) cell s9 fractions
In summary, the present data demonstrate that IPH is a poor substrate of CES1. Nevertheless, albeit at a markedly lower rate, IPH was predominantly metabolized (i.e., hydrolyzed) by CES1, with little to no contribution from CES2, an observation similar to that which is known to be the case with MPH and established structure-activity relations for hCES1 and its ester substrates (Ross and Crow 2007).
Transesterification potential of IPH versus MPH
MPH was efficiently converted to EPH via CES1 catalyzed transesterification in the presence of ethanol, with a velocity of 423.3±44.4 pmole/min/mg protein. In contrast, IPH displayed significant resistance to CES1-mediated transesterification, exhibiting a velocity of 47.5±3.3 pmole/min/mg protein (Fig. 3). Furthermore, no EPH formation was observed following the incubation of MPH and IPH with ethanol when CES1 was not present. In addition to yielding the novel transesterification metabolite EPH (Markowitz et al. 1999; Patrick et al., 2007), ethanol also significantly decreased the formation of the hydrolytic product RA following incubation with MPH.
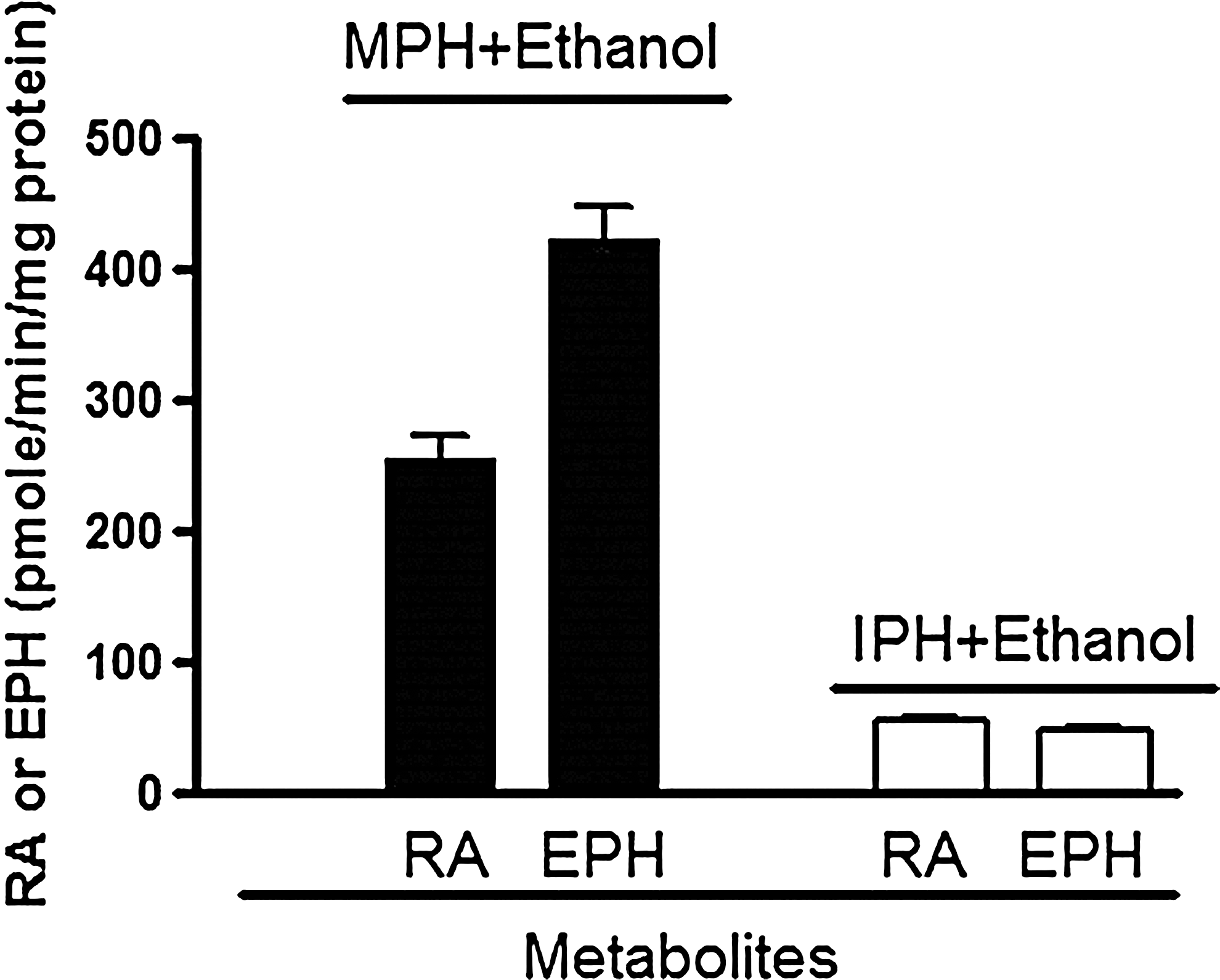
Carboxylesterase 1 (CES1) catalyzed hydrolysis and transesterification of methylphenidate (MPH) and isopropylphenidate (IPH) in the presence of ethanol. The hydrolytic metabolite ritalinic acid (RA) and the transesterification product ethylphenidate (EPH) were determined after the incubation of MPH and IPH with CES1 cell s9 fractions in the presence of ethanol at 37°C for 60 minutes. Data are means from three independent experiments with error bars representing SD.
Rat locomotor activity
The time course of locomotor response over a 120 minute period following i.p. administration of IPH (10 mg/kg) versus saline is shown in Figure 4a. As expected, IPH produced robust effects on locomotor activity in rats when compared with saline injections at 10 minute intervals recorded post-dosing, with a mean of nearly 1200 counts recorded during the initial 10 minute measurement. Also shown in Figure 4b are the cumulative locomotor activity counts over the entire 120 minute study period. Racemic IPH administration significantly elevated the cumulative locomotor activity counts in comparison with saline-injected rats (p<0.01).
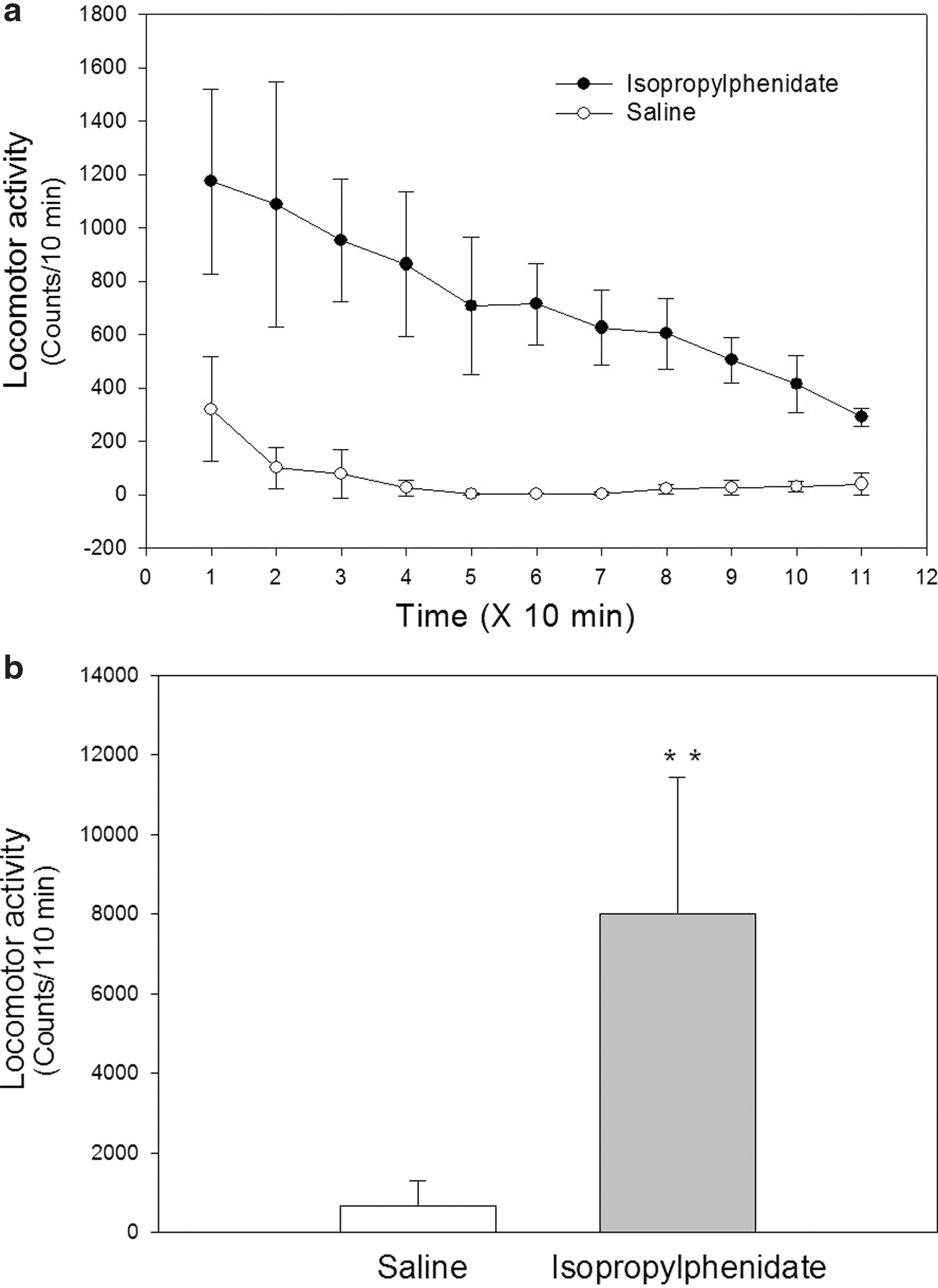
Locomotor activity at 10 minute intervals
Discussion
The present study provides a systematic assessment of in vitro and in vivo pharmacological activity, including the catalytic hydrolysis and ethanolysis rates of CES1-mediated actions using the MPH ester homolog IPH. Evaluation of the binding affinities of IPH, MPH, and EPH for DAT, NET, and SERT revealed substantial differences among these ester congeners. Overall, the transporter binding data generated for the prototype “phenidate” compound, MPH, was consistent with the majority of published in vitro reports conducted using similar methodologies (Markowitz and Patrick 2008). All tested compounds showed similar and significant binding affinities for DAT with little interaction with SERT (Table 1). With regard to NET, IPH exhibited substantially less binding affinity than MPH which was the highest of the three compounds, and significantly less than EPH which exhibited modest binding affinity at NET when these compounds were tested at a single concentration of 10 μM. The results of the complementary cellular functional studies indicated that all tested agents produce a high degree of DA uptake inhibition and few differences in these action were noted among the three (Table 2). Uptake of NE was found to be significantly lower for IPH relative to both MPH and EPH at the concentration of 10 μM. Finally, 5-HT uptake was not significantly influenced by any of the agents, although EPH appeared to exert the greatest effect of the three compounds under the present experimental conditions.
Taken together, the preliminary pharmacological screening of monoamine transporter binding and cellular uptake studies suggest that IPH is primarily a dopaminergic compound with significantly less noradrenergic activity than MPH or EPH at the concentration of 10 μM. Although there is substantial evidence of a noradrenergic component in some cases of ADHD pathophysiology as well as MPH pharmacotherapy, the prevailing view is that often clinically significant cardiovascular side effects (increased heart rate and blood pressures) associated with MPH, as well as amphetamines, are primarily mediated by the noradrenergic, sympathomimetic component of these psychostimulant actions. Accordingly, an agent that appears to have less noradrenergic activity, such as IPH, could potentially provide an improved safety profile relative to existing drugs (e.g., MPH) presently employed in ADHD treatment.
The results of enzymatic hydrolysis experiments conducted using HLM, HIM, and transfected cells overexpressing CES1 also produced substantially different results for the IPH compound relative to MPH and EPH. The data generated in the present study demonstrate that IPH is a relatively poor substrate of CES1, which is in contrast to what is observed for MPH and EPH (Figure 2). IPH was predominantly metabolized (i.e., hydrolyzed) by CES1 to RA, an inactive metabolite common to all three compounds, at a significantly lower rate relative to MPH and EPH. There was little to no contribution to IPH metabolism by CES2.
The pharmacologically active metabolite d-EPH can be formed in subjects co-ingesting MPH and ethanol, but it is not formed to the extent that the inactive l-EPH is formed (Patrick et al. 2005b, 2007). Co-abuse of MPH and alcohol has been well documented, and the recognized interaction between MPH and ethanol has become an area of concern in view of the associated elevation of d-MPH by ethanol (Patrick et al., 2007). Our in vitro incubation studies revealed that IPH displays great resistance to CES1-mediated transesterification, indicating that IPH has significantly reduced interaction potential when co-ingested with ethanol relative to MPH (Fig. 3).
The mechanism by which IPH is less efficiently hydrolyzed or transesterified relative to MPH can be speculated as based on the bulkier isopropyl substituent providing sufficient steric hindrance in accessing the active site of CES1 relative to MPH. This is consistent with current theory on structural requirements for CES1 substrates, which generally describes molecules esterified by a small alcohol group and also containing a large acyl group (e.g., MPH) (Imai 2006). It is noted, conversely, CES2 tends to show greater catalytic activity toward structures with larger alcohol groups and smaller acyl groups (Imai 2006).
In the present investigation, i.p. administration of racemic IPH to rats produced potent locomotor activity effects, as consistent with the pharmacology of MPH and other DAT-active psychostimulants that have proven useful in the management of ADHD. These data add to existing in vitro data suggesting IPH to be a significantly active central nervous system (CNS) compound at mg/kg doses similar to those used in assessments of MPH in eliciting classic stimulant-induced behavioral responses in rodents (Patrick et al. 1987). Furthermore, when compared with the earlier study by Patrick and associates (1987) characterizing the pharmacology of MPH isomers, racemic IPH dosed at 10 mg/kg appeared to produce more potent and sustained locomotor responses than those of d-MPH dosed at 5 mg/kg.
Conclusions
The present report provides in vitro evidence that IPH displays pharmacological characteristics of a CNS stimulant with a high affinity for DAT, as well as potent effects on cellular uptake of DA. However, unlike MPH, IPH has only minor effects on NE, which could theoretically provide a more desirable safety/toxicity profile. Additionally, a substantially slower rate of enzymatic hydrolysis and transesterification via CES1 was noted relative to MPH, suggesting less potential of drug interaction with ethanol, a longer duration of action and an extended dosage interval could be utilized, which is viewed as necessary in the current treatment of ADHD with MPH. Finally, in vivo studies demonstrated potent stimulating effects on locomotor activity in IPH dosed rats similar to that typically observed following MPH or amphetamine dosing. Forthcoming dose-response and time course in vivo activity assessments incorporating direct comparisons with MPH and expanded in vitro investigation of monoamine activities (see Patrick et al. 2005b) will further advance the present preclinical investigations, setting the stage for potential new drug development.
Clinical Significance
MPH exhibits a relatively short half-life as a result of rapid hydrolytic metabolism catalyzed by hepatic CES1, which necessitates multiple daily doses, or the use of expensive MR formulations to achieve symptom control throughout the day. Additionally, significant inter-individual variability in response to MPH therapy has been consistently documented, which is, at least in part, because of varied CES1 expression and activity caused by genetic polymorphisms and environmental factors. The present study revealed that IPH is more resistant to CES1-catalyzed hydrolysis and transesterification reactions than MPH, indicating that IPH may offer a longer duration of action and less potential for drug–drug interactions via CES1, and that it will be the subject of addition preclinical studies.
Footnotes
Acknowledgment
The authors thank Dr. George R. Breese, University of North Carolina-Chapel Hill, in whose laboratory the locomotor activity study was conducted.
Disclosures
Dr. Patrick has been a consultant and expert witness with Johnson & Johnson, and a consultant for Celgene, Janssen-Ortho, Novartis, Noven, and UCB. Drs. Markowitz and Zhu have no conflict of interests to declare.