
Abstract
Objective:
Quetiapine is an atypical antipsychotic with demonstrated efficacy in the treatment of adolescent schizophrenia and pediatric bipolar mania. Large, placebo-controlled studies of interventions in pediatric bipolar depression are lacking. The current study investigated the efficacy and safety of quetiapine extended-release (XR) in patients 10–17 years of age, with acute bipolar depression.
Methods:
This multicenter, double-blind, randomized, placebo-controlled study investigated quetiapine XR (dose range, 150–300 mg/day) in pediatric outpatients with an American Psychiatric Association, Diagnostic and Statistical Manual of Mental Disorders, 4th ed., Text Revision (DSM-IV-TR) diagnosis of bipolar I or bipolar II disorder (current or most recent episode depressed) treated for up to 8 weeks (ClinicalTrials.gov identifier: NCT00811473). The primary study outcome was mean change in Children's Depression Rating Scale–Revised (CDRS-R) total score. Secondary efficacy outcomes included CDRS-R-based response and remission rates.
Results:
Of 193 patients randomized to treatment, 144 patients completed the study (75.3% of quetiapine XR group [n=70]; 74.0% of placebo group [n=74]). Least squares mean changes in CDRS-R total score at week 8 were: −29.6 (SE, 1.65) with quetiapine XR and −27.3 (SE, 1.60) with placebo, a between-treatment group difference of −2.29 (SE, 1.99; 95% CI, −6.22, 1.65; p=0.25; mixed-model for repeated measures analysis). Rates of response and remission did not differ significantly between treatment groups. The safety profile of quetiapine XR was broadly consistent with the profile reported previously in adult studies of quetiapine XR and pediatric studies of quetiapine immediate-release (IR). Potentially clinically significant elevations in clinical chemistry values included triglycerides (9.3%, quetiapine XR; 1.4%, placebo group) and thyroid stimulating hormone (4.7%, quetiapine XR; 0%, placebo group). An adverse event potentially related to diabetes mellitus occurred in 3.3% of the quetiapine XR versus no adverse events in the placebo group.
Conclusions:
Quetiapine XR did not demonstrate efficacy relative to placebo in this 8 week study of pediatric bipolar depression. Quetiapine XR was generally safe and well tolerated.
Introduction
B
The immediate-release (IR) formulation of quetiapine has proven efficacy in the treatment of adults with schizophrenia, bipolar mania, and bipolar depression (Buckley 2004; Vieta et al. 2005; Thase et al. 2006; Calabrese et al. 2005; McElroy et al. 2010; Young et al. 2010). Recently, quetiapine IR demonstrated significant efficacy versus placebo in a 3 week trial of youth 10–17 years of age with acute bipolar mania (n=277; dose, 400 or 600 mg/day) and in a 6 week study of adolescents 13–17 years of age with acute schizophrenia (n=220; dose, 400 or 800 mg/day) (Findling et al. 2013; Pathak et al. 2013). Quetiapine IR was generally well tolerated in these pediatric studies, with a similar safety profile to that observed in adult studies. A 26 week, open-label, continuation study that included patients from these acute bipolar mania and schizophrenia trials confirmed that quetiapine, flexibly dosed at 400–800 mg/day, was generally safe and well tolerated (Findling et al. 2013). A previous 8 week, pilot study of quetiapine IR in adolescents with bipolar depression (n=32) reported efficacy outcomes for quetiapine that did not differ significantly from placebo, indicating the need for a larger, controlled trial of quetiapine in pediatric bipolar depression (DelBello et al. 2006, 2007, 2009).
An extended-release (XR) formulation of quetiapine has been developed that provides a longer time to peak concentration and a lower peak plasma concentration when compared with the IR formulation. Quetiapine XR, in addition to efficacy as monotherapy in adults with schizophrenia, bipolar mania, and bipolar depression, has efficacy as monotherapy in adults with generalized anxiety disorder and as both monotherapy and adjunctive therapy in adults with major depressive disorder (MDD) (Kahn et al. 2007; Weisler et al. 2009; Bandelow et al. 2010; Bauer et al. 2010; Suppes et al. 2010; Cutler et al. 2011).
The current placebo-controlled study investigated the efficacy and safety of quetiapine XR monotherapy (flexibly dosed between 150 and 300 mg/day) in patients 10–17 years of age with acute bipolar depression.
Methods
Study design
This was an 8-week, double-blind, randomized, parallel-group, placebo-controlled study conducted at 42 centers in seven countries (Colombia, India, Mexico, Serbia, South Africa, Taiwan, and the United States) between January 27, 2009 and November 1, 2010 (Study D144AC00001; ClinicalTrials.gov identifier: NCT00811473). The study consisted of a screening and enrollment period of up to 35 days (including washout), an 8 week double-blind treatment period of quetiapine XR or placebo, and a 1 week safety follow-up period.
The study was performed in accordance with the ethical principles of the Declaration of Helsinki and was consistent with the International Conference on Harmonization (ICH)/Good Clinical Practice (GCP). All patients provided informed assent and their parents or legal guardians provided informed consent before study entry. Patients and their parents or guardians could request withdrawal from the study at any time. Significant worsening of the condition, indicated by a Children's Depression Rating Scale – Revised (CDRS-R) item 13 score of >3 or a Clinical Global Impressions for Bipolar Disorder-Change (CGI-BP-C) score of ≥6 at day 15 or later, was a criterion for study discontinuation.
Patients
Boys and girls 10–17 years of age with a Diagnostic and Statistical Manual of Mental Disorders, 4th ed., Text Revision (DSM-IV-TR) diagnosis of bipolar I or bipolar II disorder (current or most recent episode depressed; duration ≥4 weeks), confirmed by the Schedule for Affective Disorders and Schizophrenia for School-Age Children-Present and Lifetime Version (K-SADS-PL), were included in the study (Kaufman et al. 1997; American Psychiatric Association 2000). Eligible patients were required to have a CDRS-R total score ≥45 and a Young Mania Rating Scale (YMRS) total score ≤16 at screening and randomization (baseline), after a 7–28 day washout of all current psychotropic medications (with the exception of psychotropics in patients with attention-deficit/hyperactivity disorder [ADHD]; described subsequently) (Young et al. 1978; Poznanski et al. 1979; Poznanski and Mokros 1996). Patients with rapid cycling, defined as four or more episodes per year, and a secondary diagnosis of comorbid ADHD, were permitted to enter the study.
Exclusion criteria were a current DSM-IV-TR Axis I disorder other than bipolar I or bipolar II depression or ADHD; a YMRS total score >16 at screening or baseline and/or criteria for bipolar disorder, most recent episode mania/hypomania/mixed, as determined by the K-SADS-PL; a history of nonresponse to adequate treatment with more than two antidepressants during the current episode or of treatment noncompliance; use of valproate within 3 days, an antipsychotic, other mood stabilizer, antidepressant, anxiolytic, hypnotic, or other psychoactive drug within 7 days, or fluoxetine within 28 days before baseline; a requirement for psychotherapy during the study period, unless initiated at least 3 months before; being a current serious suicidal or homicidal risk, a CDRS-R item 13 score of ≥3 at enrollment or randomization, or a suicide attempt within 6 months prior to enrollment; or clinically significant deviations from normal reference ranges of clinical laboratory parameters.
Study medication
The dose of quetiapine XR investigated (range, 150–300 mg/day) is consistent with the dose investigated in adults with acute bipolar depression (Suppes et al. 2010). Patients were randomly assigned as outpatients to receive in a 1:1 ratio either quetiapine XR or placebo, orally, once daily, in the evening, within each of two age strata (i.e., children 10–12 years of age or adolescents 13–17 years of age). Doses of study medication were titrated in 50 mg increments, starting at 50 mg on day 1, 100 mg on day 2, and 150 mg on day 3. In cases of clinical deterioration at the 2 or the 3 week visit, defined as a CGI-BP-C score ≥5, the daily dose of study medication could be increased in stepwise manner up to 300 mg at the treating physician's discretion. Other patients who displayed deterioration or no improvement (i.e., CGI-BP-Severity of Illness [CGI-BP-S] score ≥4) after 4 or more weeks could receive an increase in dose up to 300 mg/day in a stepwise manner. If unacceptable tolerability occurred, investigators had the discretion to reduce the study medication dose within the range of 150–300 mg/day. If a 150 mg dose was not tolerable, the patient was withdrawn from the study.
Concomitant treatment with psychostimulants (centrally acting sympathomimetics, including amphetamine, dexamphetamine, and methylphenidate) was permitted in patients with ADHD if the prescribed dose had been stable for ≥30 days prior to baseline. No psychostimulant dose adjustment was allowed during the study. Nonpsychoactive medications considered to be necessary for the patient's well-being were permitted during the study at the discretion of the investigator. Adjunctive medications were not permitted for extrapyramidal symptoms (EPS).
Primary study outcome
The primary study outcome was the mean change in CDRS-R total score from baseline to end-point in patients treated with quetiapine XR or placebo. The CDRS-R is a 17 item, clinician-administered scale used to assess the severity of depression in children and adolescents, including changes in the severity of depression in response to treatment. CDRS-R total scores can range from 17 to 113, with a higher total score indicative of more severe depression (Poznanski et al. 1979, 1996). CDRS-R scores were measured at screening and baseline and at weekly visits up to week 8.
Secondary study outcomes
Secondary study outcomes included response and remission rates. Response was defined as a ≥50% reduction from baseline in the CDRS-R total score (adjusted for non-zero baseline score by subtracting 17 from baseline and postbaseline scores) and remission was defined as a CDRS-R total score ≤28 in patients who were assessed at week 8. Changes in CGI-BP-S and CGI-BP-C scores (Spearing et al. 1997) were also assessed.
Safety and tolerability assessments included treatment-emergent adverse events (TEAEs) reported any time during the study and for up to 30 days after the last medication dose, coded using the Medical Dictionary for Regulatory Activities (MedDRA). The AEs that were assessed included suicidality, EPS (including incidences of akathisia, akinesia, Parkinsonism, restlessness, tardive dyskinesia, and tremor), AEs potentially related to diabetic events (gestational diabetes, hyperglycemia, hyperinsulinemia, insulin resistance, and non-insulin-dependent diabetes mellitus), QT prolongation, somnolence, and neutropenia/agranulocytosis. Movement disorders were assessed as AEs and by Simpson–Angus Scale (SAS), Barnes Akathisia Rating Scale (BARS), and Abnormal Involuntary Movement Scale (AIMS) scores. Emergent mania or hypomania was assessed by a YMRS score ≥16 on two consecutive assessments or at final visit. Suicidality was assessed utilizing standardized classifications similar to those in the Columbia-Suicide Severity Rating Scale (C-SSRS) (Posner et al. 2007, 2011), with potential classification into six suicidal behavior and five suicidal ideation types. Clinical chemistry and hematology assessments (undertaken by a central laboratory), vital signs, 12 lead electrocardiogram (ECG), and physical examinations were included in the safety evaluations.
Adverse events, YMRS score, C-SSRS, weight, and vital signs were reported at screening, baseline, and weekly visits up to week 8. Movement rating scales were assessed at baseline and weeks 4 and 8 (or final visit), whereas clinical chemistry, hematology, ECG, and physical examination assessments were recorded at screening and weeks 4 (excluding ECG and physical examination) and 8. An additional 2–4-week safety follow-up period for measurement of blood pressure (BP) was added as a protocol amendment for patients whose sitting systolic or diastolic BP was >95th percentile, adjusted for age and gender, at week 8 or final visit (National High Blood Pressure Education Program [NHBPEP] Working Group on Hypertension Control in Children and Adolescents 2005). Recall visits were added to the protocol for assessment of prolactin and thyroid function, when it was discovered that the central laboratory had failed to perform these tests in all patients at study completion.
Compliance with study medication was assessed by returned-tablet counts, and was calculated as the estimated number of tablets taken, divided by the number of tablets prescribed, expressed as a percentage. Patients achieving a compliance rate ≥75% and <120% were considered to be compliant. A figure >100% would be assumed to result from a failure of patients to return tablets prescribed as contingency stock to maintain access to medication in the event of delay, cancellation, or rescheduling of a study visit.
Statistical analyses
Ninety-two evaluable patients per treatment group were estimated to be required to provide at least 85% power to detect a difference of four points between the quetiapine XR and placebo groups for the mean change in CDRS-R total score based on the intent-to-treat (ITT) set analyses. This sample size calculation assumed a pooled standard deviation (SD) of 9 and a two sided test at an overall experiment type I error rate of 0.050, consistent with the published literature reporting controlled trials in adolescents with depression (Wagner et al. 2003; March et al. 2004; Wagner et al. 2004).
Efficacy analyses were conducted on the modified ITT (mITT) analysis set, which included all randomized patients who received at least one dose of study medication and had a baseline and at least one postbaseline CDRS-R assessment, and on the per-protocol (PP) analysis set, which included all mITT patients without major protocol violations. The primary study outcome of change from baseline to week 8 in the CDRS-R total score in the mITT analysis set was analyzed using a mixed-model for repeated measures (MMRM) approach. The MMRM model included baseline CDRS-R total score as covariate; age stratum, treatment group, time point, and treatment group-by-time point interaction as fixed effects; and centers and patients within treatment group as random effects. An unstructured covariance structure was used to model within-patient error, and the Kenward-Roger approximation estimated degrees of freedom. To test the robustness of the primary study analysis, analyses were repeated on the PP analysis set and on the mITT and PP sets using robust variance estimates. The primary study outcome was also analyzed using an analysis of covariance (ANCOVA) model with a last observation carried forward (LOCF) approach, including baseline CDRS-R total score as covariate, age stratum and treatment group as fixed effects, and centers as random effect.
Generalized estimating equations (GEE) were used to assess treatment group differences in response and remission rates at week 8. Factors included age stratum, treatment group, visit, treatment group-by-visit interaction, and baseline CDRS-R total score. Changes in CGI-BP-S and CGI-BP-C scores were analyzed using MMRM analysis, including baseline CGI-BP-S score as covariate; age stratum, treatment group, time point, and treatment group-by-time point interaction as fixed effects; and centers and patients within treatment groups as random effects.
Safety variables were evaluated on the safety analysis set, which included all randomized patients who received at least one dose of the study medication, and are presented by treatment group using descriptive statistics.
All statistical comparisons were based on a two sided test using an α level of significance of 5%. Analyses were run using SAS® software, version 9.2 or higher (SAS Institute, Cary, NC).
Results
Of 262 patients enrolled in the study, 193 patients (n=93, quetiapine XR group; n=100, placebo group) were randomized to study treatment (Fig. 1). Major reasons for nonrandomization included a failure to meet all the study criteria (n=56) and voluntary discontinuation by the patient or parent/guardian (n=11). The most common reasons for failure to meet study criteria included: Failure to provide a CDRS-R total score ≥45 at screening and randomization (n=14), failure to meet DSM-IV-TR criteria for bipolar I or bipolar II disorder (current or most recent episode depressed) (n=10), and posing a current serious suicidal or homicidal risk (n=6). In total, 192 patients (n=92, quetiapine XR; n=100, placebo) received at least one dose of the study medication and were included in the mITT and safety analysis sets. The PP population included 134 patients (n=63, quetiapine XR; n=71, placebo). The most common reason for exclusion from the PP population was noncompliance (i.e., <75% compliance) with study medication during the entire study period (n=22, quetiapine; n=25, placebo). Eighty-nine of the 193 randomized patients attended the recall visit for assessment of prolactin and thyroid function, at between ∼3 and 8 months after study discontinuation (n=46, quetiapine XR; n=43, placebo).
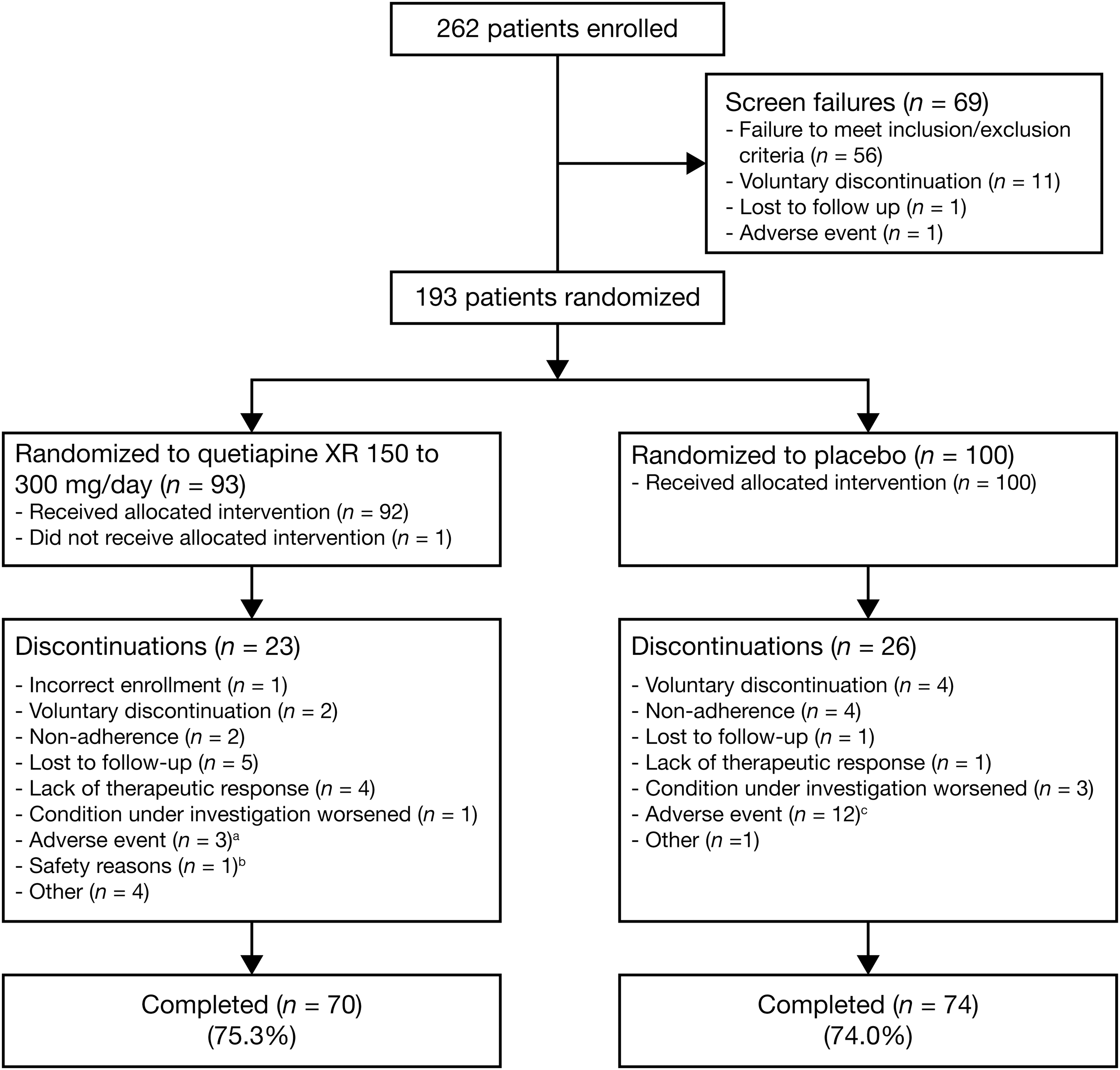
Disposition of youth with bipolar depression in the quetiapine (150–300 mg/day) and placebo groups. aDiscontinued because of: headache, sedation, and increased fatigue (n=1); somnolence (n=1); and a serious adverse event of agitation (n=1). bAssessed by investigator; no further information available. cDiscontinued because of: irritability (n=3; one of whom also reported headache); somnolence (n=2); sedation, neutropenia, and neutrophil count decreased (n=1, each); and serious adverse events of aggression, social stay hospitalization, exacerbation of bipolar I symptoms, and exacerbation of depressive symptoms (n=1, each).
Baseline demographic characteristics were similar between the quetiapine XR and placebo groups (Table 1). A total of 84 patients (43.8%) had comorbid ADHD, including 38 patients (41.3%) in the quetiapine XR group and 46 patients (46.0%) in the placebo group; 47 patients with ADHD (24.5% of the study population) were taking psychostimulants, including 20 patients (21.7%) in the quetiapine XR group and 27 patients (27.0%) in the placebo group. Baseline CDRS-R total scores were 61.6 (SD, 9.9) in the quetiapine XR and 60.1 (SD, 9.0) in the placebo group.
XR, extended release; SD, standard deviation; BMI, body mass index.
The study was completed by 144 patients, including 70 patients (75.3%) in the quetiapine XR and 74 patients (74.0%) in the placebo group. Adverse events were the most common reason for study discontinuation in the total study population (3.2%, quetiapine XR; 12.0%, placebo) (Fig. 1).
Quetiapine XR was administered at a mean modal dose of 204.9 mg/day. One or more concomitant medications were taken by 59.8% of the quetiapine XR and 64.0% of the placebo group.
Efficacy outcomes
Primary efficacy outcome
Mean CDRS-R total scores decreased from baseline to week 8 in both the quetiapine XR and placebo groups (Fig. 2). Least squares (LS) mean changes in CDRS-R total score in the mITT analysis set were −29.6 (SE, 1.65) with quetiapine XR and −27.3 (SE, 1.60) with placebo, a between-treatment group difference of −2.29 (SE, 1.99; 95% CI, −6.22, 1.65; p=0.25; MMRM).
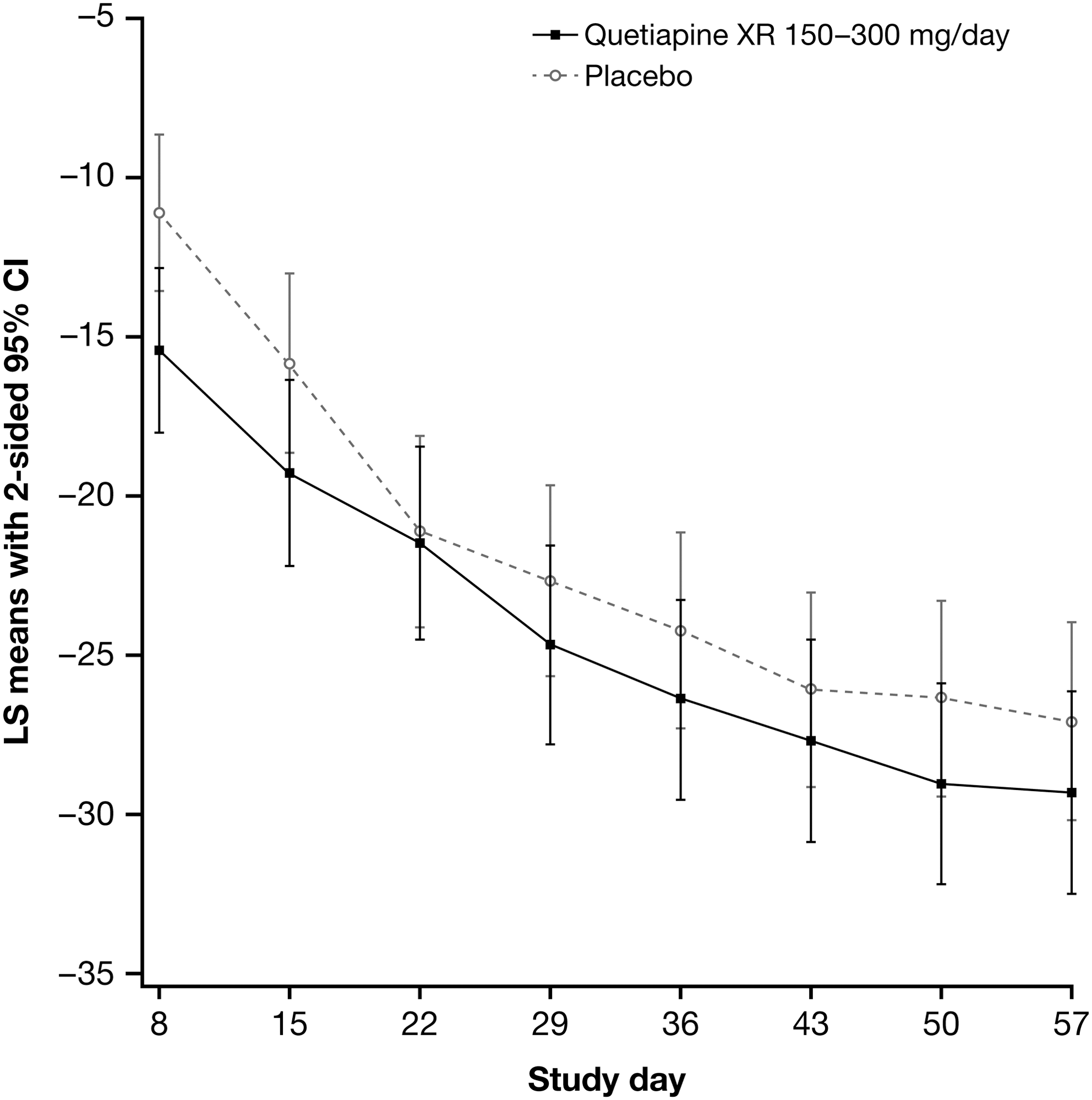
Least squares (LS) mean change from baseline in Children's Depression Rating Scale–Revised (CDRS-R) total score (with 95% CI) at each visit by treatment group (mixed-model for repeated measures [MMRM] analysis; modified ITT [mITT] analysis set).
Supportive analyses to test the robustness of the primary analysis were consistent with the primary efficacy findings, including MMRM analysis of the PP set (p=0.26, between-treatment group comparison), MMRM modeling of the mITT set with robust variance estimates (p=0.26), and ANCOVA modeling with LOCF approach (p=0.18).
Subgroup analyses of the mITT set by MMRM modeling provided results consistent with the primary analysis for patients with or without rapid cycling; with bipolar I or bipolar II disorder; and 10–12 years or 13–17 years of age. Also in agreement with the primary efficacy analysis were subgroup analyses of patients without comorbid ADHD, patients with comorbid ADHD who were not taking concomitant psychostimulants, and patients with comorbid ADHD who were taking concomitant psychostimulants (Table 2).
CDRS-R, Children's Depression Rating Scale–Revised; MMRM, mixed-model for repeated measures; mITT, modified intent-to-treat; XR, extended release; SE, standard error; ADHD, attention-deficit/hyperactivity disorder.
Response and remission rates
Rates of response (i.e., ≥50% reduction in adjusted CDRS-R total score from baseline to week 8, observed cases) were 63.0% with quetiapine XR (n=58) and 55.0% with placebo (n=55) (odds ratio [OR] 1.20; p=0.63). Rates of remission (i.e., CDRS-R score ≤28 at week 8, observed cases) were 45.7% with quetiapine XR (n=42) and 34.0% with placebo (n=34) (OR, 1.60; p=0.16).
CGI-BP-S and CGI-BP-C score changes
Least squares (LS) mean changes in CGI-BP-S score from baseline to end-point were not significantly different between the quetiapine XR and placebo groups (−1.72 vs. −1.35; between-group difference −0.38; p=0.05). LS mean changes in CGI-BP-C score from baseline to end-point also did not differ significantly (2.4 vs. 2.6; between-group difference −0.19; p=0.33). A higher proportion of patients treated with quetiapine XR had a CGI-BP-C classification of “much improved” or “very much improved” at end-point (52.2%) when compared with placebo (40.0%), but this difference was not significant (OR, 1.51; p=0.20).
Safety outcomes
TEAEs were reported by 68 (73.9%) of the quetiapine XR and 66 (66.0%) of the placebo group. The most common TEAEs in the quetiapine XR group were headache, sedation, dizziness, somnolence, diarrhea, fatigue, and nausea (Table 3). Three patients (3.3%) in the quetiapine XR group and 12 patients (12.0%) in the placebo group discontinued the study because of AEs. Serious AEs were reported by one patient (agitation) in the quetiapine XR group and by four patients (aggression, social stay hospitalization, exacerbation of bipolar I symptoms, and exacerbation of depressive symptoms) in the placebo group.
XR, extended release.
One (1.1%) patient in the quetiapine XR group experienced a TEAE of self-injurious behavior potentially associated with suicidality; this patient was also identified in the post-hoc C-SSRS analysis. The injurious behavior was assessed as nonserious and unrelated to study medication. Post-hoc C-SSRS analysis identified the occurrence of suicidal ideation in six patients in the quetiapine XR group and eight patients in the placebo group, the emergence of suicidal ideation (i.e., without suicidal ideation at baseline) in five and six patients in the respective groups, and a worsening of ideation in one patient in the placebo group versus none in the quetiapine XR group. There were no cases of suicidal behavior in either group.
One patient (1.1%) in the quetiapine XR group had an AE potentially related to EPS (restlessness), compared with none in the placebo group. Mean (SD) changes from baseline to end of treatment in SAS scores were: 0.0 (0.87) in the quetiapine group and 0.0 (0.81) in the placebo group. Respective changes in AIMS score were −0.1 (0.33) and 0.0 (0.82), and changes in BARS score were −0.1 (0.29) and 0.0 (0.71). Patients were classified as showing an improvement or a worsening in SAS, BARS, and AIMS score at the end of treatment, based on a ≤1 point or ≥1 point change in score, respectively; all other patients were classified as showing no change. The majority of patients in both groups showed either no change or an improvement in SAS, BARS, and AIMS score during the study. For example, SAS score was unchanged in 80.4% of the quetiapine XR and 78.0% of the placebo group, improved in 10.9% and 12.0%, and worsened in 7.6% and 5.0%, respectively.
Three patients (3.3%) in the quetiapine XR group had an AE potentially related to diabetes mellitus (n=2, thirst; n=1, new onset diabetes mellitus), compared with none in the placebo group. No adverse events were considered potentially related to QT prolongation. Among other AEs of special interest, 13 patients (14.1%) in the quetiapine XR and 11 patients (11.0%) in the placebo group experienced an AE potentially related to somnolence, whereas three patients (3.0%) in the placebo group experienced an adverse event potentially related to neutropenia (n=2) or neutrophil count decreased (n=1), compared with none in the quetiapine XR group. Treatment-emergent mania or hypomania, assessed by YMRS score ≥16, was reported in two (2.2%) patients in the quetiapine XR group and 10 (10.0%) patients in the placebo group.
Mean changes in clinical chemistry parameters indicated no marked differences between the quetiapine XR and placebo groups from baseline to the end of treatment or recall visit (Table 4). With the exception of triglycerides and thyroid stimulating hormone, the proportions of patients with potentially clinically significant shifts in clinical chemistry, hematology, and vital sign values were generally low and similar between the treatment groups (Tables 5, 6, and 7). At the end of treatment, mean (SD) weight gain was 1.3 (2.14) kg in the quetiapine and 0.6 (2.39) kg in the placebo group; 11 (12.5%) of the quetiapine XR and 6 (6.0%) of the placebo group experienced weight gain ≥7%.
n, number of patients with available data at baseline and end-of-treatment assessments.
Values assessed at recall visit (3–8 months after study discontinuation).
n, number of patients with available data at baseline and recall assessments.
XR, extended release; SD, standard deviation.
N, number of patients with available data at baseline and week 8 assessments.
Number of patients with available data at baseline and recall assessments.
Values assessed at recall visit.
XR, extended release; NA, not applicable (no potentially clinically significant values defined); LLN, lower limit of normal; ULN, upper limit of normal.
N, number of patients with available data at baseline and week 8 assessments.
XR, extended release; NA, not applicable (no potentially clinically significant values defined).
N, number of patients with available data at baseline and end-of-treatment assessments.
SBP, systolic blood pressure; DBP, diastolic blood pressure; bpm, beats per minute.
Measurement of sitting systolic or diastolic BP was added as a protocol amendment at 6 months before the end of the study, with the objective of providing 2–4-week follow-up on patients whose sitting systolic or diastolic BP was above the 95th percentile at final visit. In the 13 patients who had sitting blood pressure measured, mean (SD) change in systolic/diastolic blood pressure at end of treatment was 7.8 (5.85)/1.2 (5.64) mm Hg in the quetiapine (n=6) and −3.6 (5.91)/–1.1(7.90) mm Hg in the placebo (n=7) group. Of these 13 patients, 1 patient in the quetiapine group had a sitting systolic blood pressure value above the 95th percentile.
Discussion
This 8 week, placebo-controlled study of quetiapine XR is the first large, controlled trial of any pharmacologic intervention in youth with acute bipolar I or II depression. The magnitude of improvement in the symptoms of depression, measured by change in CDRS-R score, was similar in the quetiapine XR and placebo groups in primary, mITT set analyses. This outcome is considered robust, as the results were supported by the PP set and other efficacy analyses. Secondary efficacy measures also supported the primary efficacy findings, as there were no significant between-group differences in CDRS-R-derived response and remission rates, or in changes in CGI-BP-S or CGI-BP-C scores.
In contrast to the current study of youth with bipolar depression, quetiapine XR has demonstrated efficacy in the acute treatment of adults with bipolar depression or MDD (Weisler et al. 2009; Bauer et al. 2010; Suppes et al. 2010). In particular, an 8-week, placebo-controlled study of adults with bipolar depression showed significant efficacy for quetiapine XR (300 mg/day) versus placebo, measured by Montgomery-Asberg Depression Rating Scale (MADRS) score, with divergence from the first week of treatment (Suppes et al. 2010). Consistent with the current study, a pilot, 8 week, placebo-controlled study of quetiapine IR (300–600 mg/day) in youth with bipolar I depression (n=32) reported no significant differences in efficacy between the treatment groups, assessed by CDRS-R total score change and response and remission rates (DelBello et al. 2009).
The high placebo response rate in this study impacts interpretation of the findings. The placebo response rate in this study (55%, assessed by ≥50% reduction in adjusted CDRS-R total score) contrasts with the placebo response rate in adult studies of quetiapine in bipolar depression (e.g., 43%, assessed by ≥50% reduction in MADRS total score, in Suppes et al. (2010). A high placebo response rate in youth with bipolar depression was also reported in the quetiapine IR study of DelBello et al. (2009). High placebo response rates have similarly been reported in trials of antidepressants in youth with MDD, in which mean placebo response rates of 48% compared with 59% for active treatments (Bridge et al. 2009; Emslie 2009). Variability in the placebo response rate has largely explained the difference between “positive” and “negative” trials of medications in MDD (Bridge et al. 2009).
Methodological factors and disease characteristics may contribute to the high placebo response rate reported in youth with depressive symptoms. Potential contributors to the high placebo response rate discussed in previous publications include the baseline level of disease severity, the duration of depressive symptoms, the impact of comorbidities, and methodological issues such as the number of participating study centers, the rating scales adopted, and the duration of lead-in period (Bridge et al. 2009; DelBello et al. 2009). A distinct difference in treatment response between youth-onset and adult-onset bipolar depression has also been postulated (DelBello et al. 2009). These conjectures emphasize the importance of a greater understanding of the role of methodology, including the impact of patient development in studies on the management of bipolar disorder.
Quetiapine XR was generally well tolerated and was associated with low rates of treatment discontinuation in this study. Mean triglyceride levels increased in youth receiving quetiapine XR in this study, as also reported in youth with bipolar mania or schizophrenia treated with quetiapine IR (Findling et al. 2013; Pathak et al. 2013). No other clinically meaningful between-group differences were observed for clinical chemistry, hematology, vital signs, ECG, or physical finding assessments. The safety profile of quetiapine XR in these youth was broadly consistent with the profile reported previously in adult studies of quetiapine XR and in pediatric studies of quetiapine IR (Suppes et al. 2010; Findling et al. 2013; Pathak et al. 2013). It is recommended that fasting blood lipid levels and BP are measured at the beginning and at regular intervals during treatment with quetiapine, whereas monitoring of fasting blood glucose tests is recommended in patients with risk factors for diabetes mellitus (Seroquel XR 2013).
Limitations of the study include the high placebo response rate that impacts interpretation, and the absence of patient- or parent-reported outcome measures. Strengths of the study include a controlled design that investigated a well-defined population of youth with bipolar depression.
Decisions on the appropriate pharmacologic management of bipolar depression in youth remain challenging, given the lack of adequately sized, controlled trials. Age-specific open-label trials have described improvements in depressive symptoms from baseline with the mood stabilizers lithium, extended-release carbamazepine, lamotrigine, and divalproex monotherapy in diverse patient groups (Patel et al. 2006; Biederman et al. 2010; Joshi et al. 2010). Methodologically rigorous controlled studies are awaited to confirm the efficacy of those mood stabilizers. Placebo-controlled evidence for the efficacy of other atypical antipsychotics, in addition to quetiapine, for the treatment of depressive symptoms in pediatric bipolar disorder, is also limited (Tohen et al. 2007).
Conclusions
Quetiapine XR (150 to 300 mg/day) did not demonstrate efficacy relative to placebo in this large, 8 week, randomized study of youth with bipolar I or II depression. These observations contrast with the efficacy of quetiapine XR demonstrated in adults with bipolar depression or MDD. Consistent with studies in adults, quetiapine XR at the dose range investigated was generally safe and well tolerated in these pediatric patients.
Clinical Significance
Understanding the disease course and having an awareness of appropriate treatment options are key to reducing the disease burden in youth with bipolar disorder. The current 8 week, double-blind, randomized study of quetiapine XR (dose range, 150–300 mg/day) is the first large (n=193 patients), multicenter, placebo-controlled trial of any intervention in youth (ages 10–17 years) with acute bipolar depression. Both quetiapine XR and placebo were associated with improvement in depressive symptoms, with no significant between-group difference. The results of this study may inform clinical practice as well as the conduct of future randomized trials in youth with bipolar depression.
Footnotes
Acknowledgment
We thank Bill Wolvey from PAREXEL, who provided medical writing support funded by AstraZeneca.
Disclosures
Dr. Findling receives or has received research support from, or acted as a consultant and/or served on a speaker's bureau for Alexza Pharmaceuticals, American Psychiatric Press, AstraZeneca, Bracket, Bristol-Myers Squibb, Clinsys, Cognition Group, Dana Foundation, Forest, GlaxoSmithKline, Guilford Press, Johns Hopkins University Press, Johnson & Johnson, KemPharm, Lilly, Lundbeck, Merck, NIH, Novartis, Noven, Otsuka, Oxford University Press, Pfizer, Physicians Postgraduate Press, Rhodes Pharmaceuticals, Roche, Sage, Seaside Pharmaceuticals, Shire, Stanley Medical Research Institute, Sunovion, Supernus Pharmaceuticals, Transcept Pharmaceuticals, Validus, and WebMD. Drs. Pathak and Liu are employees, and Dr. Earley is a former employee, of AstraZeneca Pharmaceuticals LP, USA. Dr. DelBello receives or has received research support from, or acted as a consultant, and/or served on a speaker's bureau for AstraZeneca, Brain and Behavior Research Foundation, Bristol-Myers Squibb, Eli Lilly, GlaxoSmithKline, Janssen, Johnson & Johnson, Martek, Merck & Co., National Institute on Alcohol Abuse and Alcoholism (NIAAA), National Institute on Drug Abuse (NIDA), National Institute of Mental Health (NIMH), Pfizer, Repligen, Shire, Schering-Plough, Somerset, and the Thrasher Foundation.
Martha Alzate, Instituto Colombiano del Sistema Nervioso Clínica Montserrat, Bogota, Colombia; Sarah Atkinson, Finger Lakes Clinical Research, Rochester, NY; Ashraf Attalla, Institute for Behavioral Medicine LLC, Smyrna, GA; Grant Belnap, Mountain West Clinical Trials LLC, Eagle, ID; Michael Bengston, University of South Florida, Tampa, FL; Prakash Bhatia, Synergy Escondido, Escondido, CA; Matthew Brams, Bayou City Research Ltd., Houston, TX; Hernan Dario Giraldo Castro, Hospital Mental de Antioquia, Bello, Colombia; Lydia Cohan, Meadowbrook Research Inc., Scottsdale, AZ; Rodrigo Cordoba, Centro de Investigaciones del Sistema Nervioso Limitada, Bogota, Colombia; Melissa DelBello, Psychiatric Professional Services Inc., Cincinnati, OH; Michael Feldman, Cutting Edge Research Group, Enid, OK; Robert Findling, University Hospital Case Medical Center, Cleveland, OH; Hitendra A. Gandhi, Sheth Vadilal Sarabhai General Hospital, Ahmedabad, India; Lawrence Ginsberg, Red Oak Psychiatry Associates PA, Houston, TX; Venu Gopal Jhanwar, Deva Mental Health Care, Durgakund, India; Nelson Handal, Harmonex Neuroscience Research, Dothan, AL; Paras Harshawat, Clinco, Terre Haute, IN; Willis Holloway, Cutting Edge Research Group, Oklahoma City, OK; Sendi Ismail, New Oakland Child/Adolescent & Family Center, Clinton Township, MI; Sanjiv Kumra, University of Minnesota, Minneapolis, MN; Aneta Lakic, Neurology and Psychiatry Clinic for Children and Adolescents, Belgrade, Serbia; Mark Lerman, Alexian Brothers Behavioral Health Hospital, Hoffman Estates, IL; Steven Lopez, Segal Institute for Clinical Research, Charleston, SC; Mohammad Malik, PsychCare Consultants Research, St. Louis, MO; Susan McElroy, The Research Institute, The Lindner Center of HOPE, Mason, OH; Dragan Mitrovic, Clinical Center Vojvodina, Vojvodina, Serbia; William Murphy, Psychiatric Associates, Overland Park, KS; Duong Nguyen, Woodland International Research Group LLC, Little Rock, AR; Patel Nilesh, Wharton Research Center, Wharton, TX; Alfonso Ontiveros, Instituto de Información de Investigación en Salud Mental, Monterrey, Mexico; Smiljka Popovic-Deusic, Institute of Mental Health, Belgrade, Serbia; Indla Ramasubbareddy, Vijayawada Institute of Mental Health and Neurosciences, Vijayawada, India; Robert Riesenberg, Atlanta Center for Medical Research, Atlanta, GA; Juan Schronen, Cape Trial Centre, Randburg, South Africa; Segal Scott, Scientific Clinical Research Inc., North Miami, FL; David Spiegel, Brighton Research Group LLC, Virginia Beach, VA; David Struble, Research Strategies Inc., Memphis, TN; Mustapha Syed, Pacific Institute of Medical Sciences, Bothell, WA; Juan Luis Vazquez Hernandez, Hospital Aranda de la Parra, León, Mexico; Chin-Bin Yeh, Tri-Service General Hospital, Taipei City, Taiwan; Jose Zaglul, Florida Clinical Research Center LLC, Bradenton, FL.