
Abstract
Objective:
The purpose of this study was to evaluate the efficacy and safety of duloxetine flexible dose in children (7–11 years) and adolescents (12–17 years) with major depressive disorder (MDD).
Methods:
Patients (n=337) in this 36 week study (10 week acute and 26 week extension treatment) received duloxetine (60–120 mg once daily [QD], n=117), fluoxetine (20–40 mg QD, n=117), or placebo (n=103). Measures included: Children's Depression Rating Scale-Revised (CDRS-R), treatment-emergent adverse events (TEAEs), and Columbia-Suicide Severity Rating Scale (C-SSRS).
Results:
Neither active drug (duloxetine or fluoxetine) separated significantly (p<0.05) from placebo on mean change from baseline to end-point (10 weeks) on the CDRS-R total score. There were no significant differences between the duloxetine or fluoxetine groups compared with placebo on serious AEs (SAEs), total TEAEs, or discontinuation for AE during acute treatment. There were no completed suicides or deaths, and no clinically significant electrocardiogram (ECG) abnormalities observed during the study. One fluoxetine and one duloxetine patient experienced alanine aminotransferase (ALT) three or more times the upper limit of normal, which resolved during the study. A total of 8 (7.1%) duloxetine patients, 7 (6.8%) placebo patients, and 9 (8.0%) fluoxetine patients had worsening of suicidal ideation from baseline during acute treatment. Of the patients with suicidal ideation at baseline, 15/19 (79%) duloxetine, 19/19 (100%) placebo, and 16/19 (84%) fluoxetine had improvement in suicidal ideation at end-point during acute treatment. One duloxetine and two fluoxetine patients had treatment-emergent suicidal behavior during the 36 week study.
Conclusion:
Trial results were inconclusive, as neither the investigational drug (duloxetine) nor the active control (fluoxetine) separated from placebo on the CDRS-R at 10 weeks. No new duloxetine safety signals were identified relative to those seen in adults.
Clinical Trial Registry Number:
NCT00849901
Introduction
L
This study incorporated a flexible dosing scheme for both duloxetine and fluoxetine during the acute treatment period (which included the 10 week primary study end-point), whereas the sister study (Emslie et al. 2014) used fixed doses of both drugs. It is possible that the response profile from a fixed dose study could underestimate effectiveness of medications; therefore, a flexible dosing strategy was employed for this study, and may also be more comparable to what is done in clinical practice. The dose range for duloxetine used in this study was based on results of a previous safety, tolerability, and pharmacokinetics study of duloxetine, which suggested that adjustment of the duloxetine total daily dose based on body weight or age is not warranted for pediatric patients, and that different total daily doses may not be warranted for pediatric patients relative to adults (Prakash et al. 2012). The dose range for fluoxetine used in this study was based on its approved dose in the treatment of pediatric MDD (20 mg/day) as well as additional studies that have shown evidence of clinical benefit at 40 mg/day for pediatric patients who do not respond to lower doses (March et al. 2004; Heiligenstein et al. 2006).
Patients and Methods
Study design
The protocol for this study (F1J-MC-HMCK) was filed with the United States Food and Drug Administration prior to study initiation. It included all of the methodology presented here, in addition to a complete statistical analysis plan. This study was approved by the ethics review boards for each study site, and conducted in accordance with good clinical practice guidelines. In accordance with the principles of the Declaration of Helsinki, a parent/legal representative of each study patient provided written informed consent prior to administration of any study drug or study procedures, and patients (children and adolescents) also provided assent as appropriate to participate in the study. Patients participated in the study at 65 psychiatric clinical sites in nine countries (United States, Finland, France, Germany, Slovakia, Estonia, Russia, Ukraine, and South Africa) from March 2009 to October 2011.
The primary objective of the study was to assess the efficacy of duloxetine (flexible dose) compared with placebo in the acute treatment of children (age 7–11 years) and adolescents (ages 12–17 years) who met criteria for MDD without psychotic features, single or recurrent episode, as defined in the Diagnostic and Statistical Manual of Mental Disorders, 4th ed., Text Revision (DSM-IV-TR) (American Psychiatric Association 2000). This objective was evaluated by assessing the mean change from baseline to end-point (10 weeks) on the Children's Depression Rating Scale-Revised (CDRS-R) total score between duloxetine and placebo.
This was a phase 3, randomized, double-blind, placebo- and fluoxetine-controlled study. Patients meeting entry criteria were randomly assigned 1:1:1 to either duloxetine flexible dose (60–120 mg once daily [QD]), fluoxetine flexible dose (20–40 mg QD), or placebo, via Interactive Voice Response System (IVRS).
The study design incorporated a 2–4 week screening period, a 10 week double-blind (flexible dose) placebo-controlled acute treatment period, a 26 week double-blind (flexible dose) long-term treatment period in which all patients received either duloxetine (60–120 mg QD) or fluoxetine (20–40 mg QD), and a 2 week double-blind dose tapering period. Clinic visits were scheduled weekly during the screening period, at weeks 1, 2, 4, 7, and 10 during the placebo-controlled acute treatment period, and at weeks 12, 14, 16, 20, 24, 28, 32, and 36 during the double-blind long-term treatment period.
This study was a flexible dosing study. All patients received six capsules of study drug to be taken once daily throughout the study. Patients randomized to duloxetine or fluoxetine initiated treatment with a low dose (duloxetine 30 mg QD or fluoxetine 10 mg QD) for 2 weeks, and then were automatically increased to duloxetine 60 mg QD or fluoxetine 20 mg QD at the 2 week time point. The duloxetine dose could be increased to 90 mg QD at the 4 week time point or later, and subsequently increased to 120 mg QD at the 7 week time point or later. The fluoxetine dose could be increased to 40 mg QD at the 4 week time point or at any later time point. Patients randomized to placebo remained on placebo throughout the 10 week acute treatment period, and were then automatically transitioned to duloxetine 30 mg QD for 2 weeks, with subsequent automatic escalation to 60 mg QD and flexible dosing (60, 90, 120 mg QD) thereafter. Dose decreases due to lack of tolerability were permitted; however, the lowest dose allowed after the 2 week time point was 60 mg QD for duloxetine and 20 mg QD for fluoxetine. Dose adjustments for all patients were based on investigators' assessment of tolerability as well as inadequate clinical response (CGI-Severity≥3) and were implemented in a blinded fashion via IVRS.
Selection of patients
Study participants were outpatient children (7–11 years) and adolescents (12–17 years), who met DSM-IV-TR criteria for MDD without psychotic features, had a CDRS-R (Poznanski et al. 1979, 1984) total score ≥40 and a Clinical Global Impressions of Severity (CGI-S) (Guy 1976) score ≥4 at the three screening visits. An MDD diagnosis was supported by the Mini International Neurospychiatric Interview for children and adolescents (MINI-Kid) (Sheehan et al. 2010) and was conducted by two independent evaluators, with at least one evaluator being a psychiatrist. Patients were required to be medically stable based on the physical examination, laboratory tests, and electrocardiogram (ECG) completed at the screening visits. Female patients were required to have a negative serum pregnancy test at screening. Patients were excluded from the study if they were pregnant or lactating, had a <20 kg baseline weight; current Axis I disorder (other than MDD) requiring pharmacotherapy; first-degree relative with bipolar I disorder; serious or unstable medical illness; serious suicide risk; history of substance abuse/dependence within the past year; or an unexplained positive urine drug screen. In general, use of concomitant medications having primarily central nervous system activity was not permitted during the study. Use of antidepressants, antipsychotics, anticonvulsants, anorexics, benzodiazepines, psychostimulants (excluding caffeine), and herbal preparations was strictly prohibited during the study. Episodic use of diphenhydramine and antiemetics was allowed. Limited use of narcotic analgesics to relieve pain from surgical procedures or acute injury was allowed, and patients could continue on stable doses of hormones, antihypertensives, oral hypoglycemics, and diuretics to treat stable medical conditions, provided that dosing was stable 3 months prior to enrollment in the study.
Efficacy measures
The clinician-rated CDRS-R total score (sum of all 17 items) (Poznanski et al. 1979, 1984) and CGI-S (Guy 1976) efficacy measures were collected at every visit. The CDRS-R was administered only by site personnel who underwent training on the use of the instrument and met predetermined inter-rater reliability criteria, which were evaluated during rater training sessions at the study startup meetings.
Safety assessments
Spontaneously reported adverse events (AEs), pulse, blood pressure, and weight were recorded at every visit. Pharmacokinetic samples were collected at various time points, and results will be presented elsewhere. Chemistry and hematology blood samples were collected at baseline and weeks 4, 10, 14, 20, 24, 36, or early termination with ECGs collected at baseline and weeks 10, 24, 36, or early termination. Prespecified definitions of potentially clinically significant (PCS) changes in vital signs and weight are shown in Table 2.
The Columbia-Suicide Severity Rating Scale (C-SSRS) (Posner et al. 2007, 2011) was used in this study to prospectively capture the occurrence, severity, and frequency of suicide-related thoughts and behaviors. Nonserious AEs obtained through the C-SSRS were recorded and analyzed separately, and were not recorded as AEs via the case report form unless the nonserious AE was spontaneously reported by the patient or the parent/legal guardian. Only serious AEs (SAEs) elicited through the C-SSRS were recorded as AEs via the study case report forms. All occurrences of suicidal ideation, behaviors, acts, or nonsuicidal self-injurious behavior during treatment were tabulated. These outcomes were also analyzed as treatment-emergent (new occurrence or worsening of preexisting events) based on lead-in baseline (defined for acute and extension treatment in Table 3), or improved from lead-in baseline.
Sample size
The study was powered to address the primary objective (contrast between flexible dose duloxetine and placebo on the CDRS-R total score mean change from baseline to 10 weeks). The powering of the study was based on an anticipated enrollment of 336 patients, randomized in a 1:1:1 ratio to duloxetine 60–120 mg QD, fluoxetine 20–40 mg QD, and placebo. It was assumed that ∼10% of patients would discontinue the trial prior to providing postbaseline data on the primary efficacy outcome, leaving ∼100 patients per treatment arm with postbaseline data. This sample size was estimated to have 80% power to detect an effect size of 0.40 (duloxetine efficacy relative to placebo on the CDRS-R total score) using a two group t test with a 0.05 two-sided significance level. The effect size of 0.4 was determined based on historical data for the effect size of duloxetine 60 mg QD in adult patients with MDD (Pritchett et al. 2007).
Statistical methods
All analyses were completed on an intent-to-treat (ITT) basis unless otherwise specified. An ITT analysis is an analysis of data by the groups to which patients are assigned by random allocation, even if the patient did not take the assigned treatment, did not receive the correct treatment, or otherwise did not follow the protocol. All analyses of continuous measures included randomized patients with both a baseline and at least one postbaseline value for the variable being analyzed.
Unless otherwise specified, for analyses of continuous measures, baseline was defined as the last measurement taken at, or prior to, the visit when the study period (acute or long-term treatment period) began; end-point was defined as the last nonmissing measurement for the study period of interest.
The protocol-specified primary analytic approach for assessing mean changes for all efficacy measures was the recommended (Lieberman et al. 2005; National Research Council 2010) restricted maximum likelihood (REML)-based mixed effects model repeated measures (MMRM) approach using all the longitudinal observations at each postbaseline visit. The MMRM model included the fixed categorical effects of treatment, pooled investigative site, visit, treatment-by-visit interaction, age category (children 7–11 years, adolescents 12–17 years), and age category-by-visit interaction, as well as the continuous, fixed covariates of the baseline value being analyzed, and the baseline value of the variable being analyzed-by-visit interaction. The baseline value of the variable being analyzed and baseline-by-visit interaction were included to account for the differing influence over time of the baseline score on the postbaseline scores. An unstructured covariance structure was used to model the within-patient errors. A Kenward–Roger correction (Kenward and Roger 1997) was used to estimate denominator degrees of freedom. Significance tests were based on least-squares means (LS means) using a two sided α=0.05.
Additional analyses of continuous efficacy and safety measures were also conducted using an analysis of variance (ANOVA) or an analysis of covariance (ANCOVA) model. When an ANOVA model was used, the model contained the main effects of treatment and pooled investigative site. An ANCOVA model, in general, refers to the ANOVA model with baseline values and age category (children 7–11 years, adolescents 12–17 years) added as covariates. Type III sum-of-squares for the LS mean was used for the statistical comparison of main effects using ANOVA or ANCOVA. Statistical inference for ANOVA or ANCOVA interaction terms was based on type II sum-of-squares for the LS mean. A last observation carried forward (LOCF) method was used for these analyses.
Response was defined as a 50% improvement on the CDRS-R total score after subtracting the 17 item base score (Emslie et al. 2002), and remission was defined as a CDRS-R total score ≤28. Probabilities of response and remission were estimated using a categorical MMRM approach, in which a marginal model based on a pseudolikelihood method was utilized and implemented in SAS PROC GLIMMIX. The model included the fixed categorical effects of treatment, pooled investigator, visit, and treatment-by-visit interaction, age category (7–11 vs. 12–17 years), age category-by-treatment interaction, as well as the continuous fixed covariate of baseline score and baseline-by-visit interaction. Categorical safety measures were analyzed using Fisher's exact test.
“Mean change” refers to adjusted LS mean change from MMRM, ANOVA, or ANCOVA model. Statistical comparisons between patients initially randomized to duloxetine or fluoxetine over the 36 week combined acute/extension periods were conducted. Statistical comparisons between treatment groups were not conducted for the extension period analyses, because of selection bias. In other words, only patients who completed the acute period of the study were included in the extension period analyses; therefore, patient characteristics at the beginning of the extension period were expected to be different between treatment groups because of lack of randomization.
Results
Patients
The flow of patients through the study is shown in Figure 1. The overall patient cohort was comprised of 40.1% children (7–11 years) and 59.9% adolescents (12–17 years) with approximately equal proportions of males (47.8%) and females (52.2%). The median age of randomized patients was 13.5 years. Among children (7–11 years), 53.3% were male and 46.7% were female. Among adolescents (12–17 years), 44.1% were male and 55.9% were female. Among female patients, 57.4% had reached menarche at study baseline. The majority of the patients were white (81.4%). This was an international multicenter study with most patients enrolled from the United States (41.5%) followed by Eastern Europe (33.5%), South Africa (19.9%), and Western Europe (5.0%).

Patient flow.
Among randomized patients, the mean age of first episode of MDD was 11.6 years. The mean number of previous episodes was 0.5 (median 0.0), and 71.5% of patients were experiencing a first episode of MDD. The mean CDRS-R total score at baseline was 59.4 (median 58.0) indicating moderately severe depression, which was consistent with the mean CGI-S score at baseline of 4.5 (median 4.0). There were no significant between-group differences in baseline demographics or psychiatric profile (Table 1). Approximately half of the study patients had a first-degree relative with diagnosed depression (49.2%), and a total of four patients had first-degree relatives with diagnosed bipolar disorder (all cases bipolar II).
Headache (14.8%) was the most frequently reported preexisting condition and the only preexisting condition reported by ≥10% of the patient population. Preexisting conditions reported by ≥2% and <10% of the patient population were: dysmenorrhea (females only, 6.8%), seasonal allergy (6.2%), asthma (5.9%), upper abdominal pain (5.0%), acne (4.7%), myopia (3.9%), insomnia (3.3%), constipation (3.0%), hypersensitivity (3.0%), upper respiratory tract infection (3.0%), anxiety (2.4%), allergic rhinitis (2.4%), arthralgia (2.4%), attention-deficit/hyperactivity disorder (2.4%), oppositional defiant disorder (2.4%), nausea (2.1%), enuresis (2.1%), and obesity (2.1%). Of these preexisting conditions, there were no statistically significant differences in the frequency of preexisting conditions at baseline between the active drug groups (duloxetine or fluoxetine) and the placebo group.
Dosing
During acute treatment, the last prescribed dose for duloxetine patients was a 30 mg titration dose (11.1%), 60 mg (17.1%), 90 mg (27.4%) and 120 mg (43.6%). During acute treatment, the last prescribed dose for fluoxetine patients was a 10 mg titration dose (7.7%), 20 mg (18.8%), and 40 mg (73.5%).
During extension treatment, the last prescribed dose of duloxetine for patients initially randomized to duloxetine and continued on duloxetine during extension treatment was 60 mg (14.5%), 90 mg (16.9%), and 120 mg (68.7%). The last prescribed dose for patients initially randomized to placebo and transitioned to duloxetine during extension treatment was a 30 mg titration dose (3.5%), 60 mg (49.4%), 90 mg (16.5%), and 120 mg (30.6%). The last prescribed dose for patients initially randomized to fluoxetine and continued on fluoxetine during extension treatment was 20 mg (16.5%) and 40 mg (83.5%).
Acute efficacy
Results from the primary efficacy measure of the study showed that duloxetine was not statistically different than placebo in the treatment of children and adolescents with MDD (Fig. 2). The active control (fluoxetine) also did not statistically differ from placebo (Fig. 2). Mean CDRS-R total scores at the 10 week end-point (MMRM) were: 35.0, 35.6, and 35.0 for duloxetine, fluoxetine, and placebo respectively. Mean CGI-S scores at the 10 week time point (MMRM) were: 2.7, 2.7, and 2.6 for duloxetine-, fluoxetine-, and placebo-treated patients, respectively, with no statistically significant differences for either active drug treatment group compared with placebo.
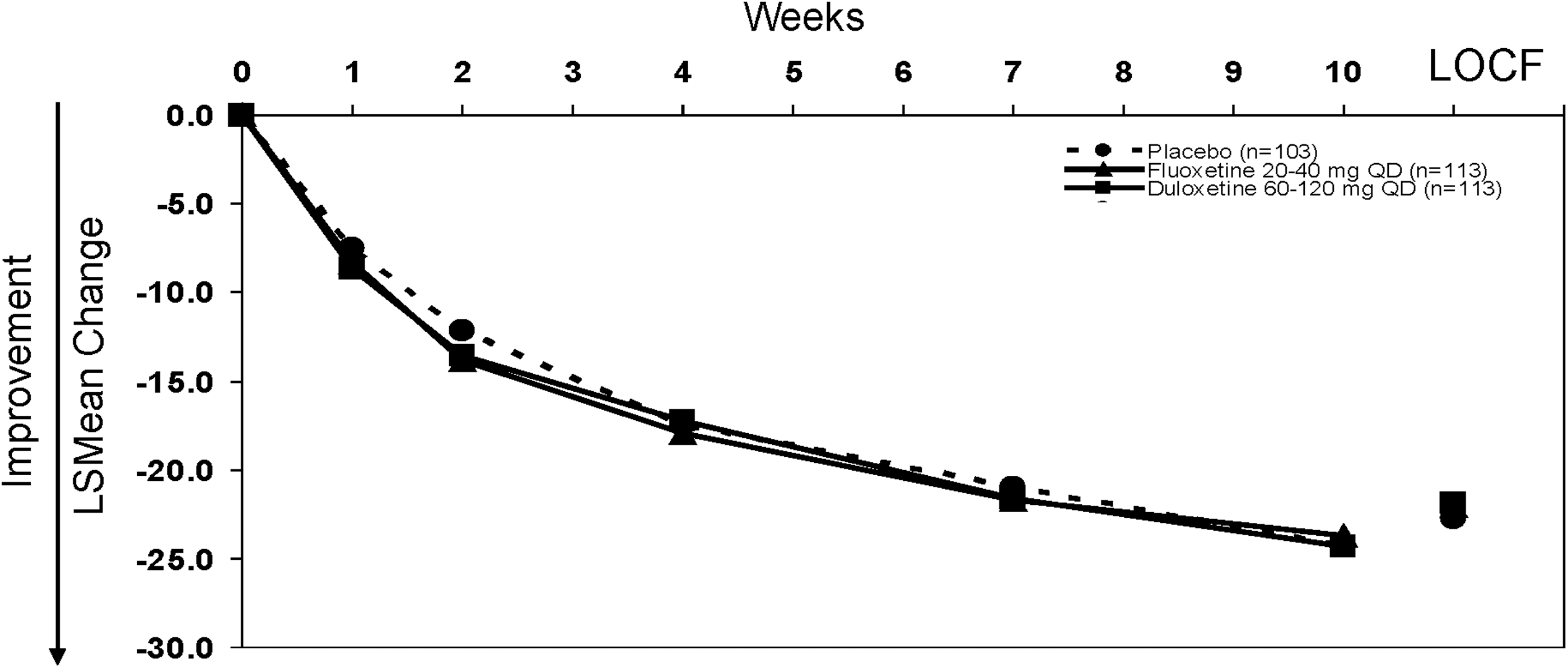
Primary outcome: Mean change on the Children's Depression Rating Scale-Revised (CDRS-R) total score from baseline to 10 weeks (mixed effects model repeated measures [MMRM]). Mean changes at 10 weeks for duloxetine-, fluoxetine-, and placebo-treated patients were −24.3, −23.7, and −24.3, respectively. Mean changes to last observation carried forward (LOCF) end-point for duloxetine-, fluoxetine-, and placebo-treated patients were −21.9, −22.0, and −22.7, respectively.
In subgroup analyses of children (7–11 years) and adolescents (12–17 years), there were no statistically significant differences in the mean change from baseline to last observation on the CDRS-R total score for duloxetine-treated children (mean change: −23.1) compared with placebo-treated children (mean change: −22.4) or for duloxetine-treated adolescents (mean change: −21.6) compared with placebo-treated adolescents (mean change: −23.2). There were also no statistically significant differences between fluoxetine-treated patients (mean change: −22.3 [children], −22.4 [adolescents]) and placebo on the CDRS-R total score in either age group. There were also no statistically significant differences in mean change from baseline to last observation on the CDRS-R total score for either active drug treatment group versus placebo in male or female subsets.
A comparison by geographic region (United States, Western Europe, Eastern Europe, and South Africa) was conducted to assess the consistency of the results among regions. Consistent with the primary study results, the treatment-by-region interaction was not statistically significant (p=0.4), suggesting consistency in the treatment response (to both drug and placebo) across all four regions.
Probabilities by mixed-effects model repeated measures (MMRM) of treatment response (50% improvement in CDRS-R total score from baseline) at 10 weeks for patients receiving duloxetine (67%) or fluoxetine (63%) did not differ significantly from that for placebo-treated patients (62%). There were also no significant differences between treatment groups in the probabilities of remission (CDRS-R total score ≤28) at 10 weeks: duloxetine 41%, fluoxetine 33%, placebo 41%.
Longer-term effectiveness
Mean CDRS-R total scores at the 36 week time point (MMRM) were: 26.0, 25.7, and 25.1 for patients initially randomized to duloxetine or fluoxetine, and patients transitioned from placebo to duloxetine, respectively. Mean CGI-S scores at the 36 week time point (MMRM) were: 1.9, 1.8, and 1.6 for patients initially randomized to duloxetine or fluoxetine, and patients transitioned from placebo to duloxetine, respectively.
For patients initially randomized to duloxetine, the probability (MMRM) of remission at 36 weeks was 72%, and for patients initially randomized to fluoxetine, it was 83%. There were no statistically significant differences in probability of remission at any time during the 36 week study between patients initially randomized to duloxetine or fluoxetine.
Safety: Patient disposition
Rates and reasons for early discontinuation during the study are shown in Figure 1. There were no statistically significant differences between the active drug treatment groups and placebo in reasons for discontinuation during the acute treatment period; however, significantly more duloxetine-treated patients discontinued because of an AE compared with fluoxetine-treated patients (7.7% duloxetine, 0.9% fluoxetine, and 2.9% placebo, p=0.019 for duloxetine vs. fluoxetine). A total of 91 fluoxetine-treated patients completed acute treatment, and 92 fluoxetine-treated patients entered the extension period, because 1 patient had discontinued from the acute period because of an AE, and was accidentally dispensed the drug. Based on ITT principle, this patient was included in the extension period analyses, although this patient did not contribute any data in the extension period.
Safety: Deaths and SAEs
No deaths occurred during the study; however, 14 patients (7 duloxetine, 6 fluoxetine, 1 placebo) experienced 20 SAEs. Of these 20 SAEs, 3 were suicide-related (1 ideation and 2 behaviors [intentional overdose and suicide attempt in one patient]) and will be discussed in the C-SSRS section. All SAEs resulted in hospitalization of the patient.
Non-suicide-related SAEs reported for duloxetine-treated patients were: drug abuse (caffeine) and panic attack (two events in one patient during acute treatment); social phobia (one patient, acute), syncope (one patient, acute), and pneumonia (one patient, extension). Non-suicide-related SAEs reported for duloxetine-treated patients after transitioning from placebo were: restlessness (one patient, extension), conversion disorder (one patient, extension), and pilonidal cyst (one patient, extension). Non-suicide-related SAEs reported for fluoxetine-treated patients were: gastritis and lymphadenitis (two events in one patient during acute treatment), ulna fracture (one patient, acute), hypomania (one patient, extension), adjustment disorder with disturbance of conduct (one patient, extension), convulsion and epilepsy (two events in one patient, extension), and spinal compression fracture (one patient during taper period). The one SAE reported for a placebo-treated patient was: major depression (one patient acute).
Safety: Adverse events
During acute treatment, the proportion of patients who experienced at least one treatment-emergent adverse event (TEAE) did not differ significantly between active drug treatment groups and placebo (duloxetine 59.8%, fluoxetine 62.4%, placebo 66.0%). During extension treatment, the proportion of patients who experienced at least one TEAE was similar for patients initially randomized to duloxetine (63.9%) and fluoxetine (62.0%), and lower than the proportion for patients transitioned from placebo to duloxetine (72.1%). The most frequently reported TEAEs (≥10%) during the study were: nausea, headache, and nasopharyngitis.
A total of 27 patients discontinued because of AEs during the 36 week study (Fig. 1), and AEs leading to discontinuation in >1 patient during the study were nausea (2 patients, duloxetine), abdominal pain upper (2 patients, 1 duloxetine and 1 placebo), depression (2 patients, duloxetine), and intentional overdose (2 patients, 1 fluoxetine [SAE, suicide attempt] and 1 placebo [not an act of self-harm, patient misunderstood how to take the study medication]).
Safety: Vital signs, weight, ECG, and laboratory results
Table 2 shows mean changes in vital signs and weight from baseline to 10 weeks (MMRM), as well as the incidence of PCS changes in weight, pulse, and blood pressure at any time and at end-point during acute treatment. In general, the incidence of PCS changes in weight, pulse, and blood pressure were lower at end-point during acute treatment as compared to at any time during acute treatment (Table 2). With regard to longer-term results, there were no statistically significant differences in blood pressure at 36 weeks between patients initially randomized to duloxetine and those initially randomized to fluoxetine. Mean weight changes at 36 weeks (MMRM) were not statistically different for patients initially randomized to duloxetine (mean increase 2.8 kg) and those initially randomized to fluoxetine (mean increase 3.0 kg). Mean change in pulse at 36 weeks was significantly (p<0.05) greater for duloxetine-treated patients (4.0 bpm) than for fluoxetine-treated patients (−1.3 bpm).
Baseline was defined as last observation during the screening period, and end-point was the 10 week end-point of the acute treatment period.
N=number of patients with high or normal values at baseline and with at least one nonmissing postbaseline measure; n=number of patients with a PCS postbaseline measurement.
N=number of patients with low or normal values at baseline and with at least one nonmissing postbaseline measure; n=number of patients with a PCS postbaseline measurement.
p<0.05 vs. placebo; § p<0.05 vs. fluoxetine.
PCS increase in blood pressure=increase ≥5mm Hg from baseline high to a value above the 95th percentile based on age, height, and sex (National High Blood Pressure Education Program Working Group 2004).
PCS decrease in pulse=decrease ≥25 bpm from baseline low to a value of <60 bpm for children or decrease ≥15 bpm from baseline low to a value <50 bpm for adolescents.
PCS increase in pulse=increase ≥15 bpm from baseline high to a value >140 bpm for children or increase ≥15 bpm from baseline high to a value >120 bpm for adolescents.
PCS decrease in weight=decrease ≥3.5% from baseline low.
Sustained elevation in blood pressure=PCS blood pressure at three consecutive postbaseline visits.
There were no statistically significant differences between patients initially randomized to duloxetine and those randomized to fluoxetine in the incidence of PCS changes in weight, pulse, or blood pressure at any time or at end-point (last observation) during the 36 week study. Among patients initially randomized to duloxetine, a total of five patients had sustained (three consecutive visits) elevation in blood pressure; three patients had sustained elevation in systolic blood pressure and four patients had sustained elevation in diastolic blood pressure (two of which also had sustained elevation in systolic blood pressure) during the 36 week study. No patients randomized to fluoxetine experienced sustained elevation in blood pressure (systolic or diastolic) during the 36 week study.
No patients in any treatment group met PCS criteria for prolonged QTc Fridericia correction (>500 msec) at any time during the study.
No patients had an SAE related to laboratory results, and no patients were discontinued because of abnormal laboratory values. One patient in the fluoxetine group had a treatment-emergent alanine aminotransferase (ALT) equal to or more than three times the upper limit of normal during the extension period, and the same patient had a treatment-emergent ALT equal to or more than five times the upper limit of normal during the extension period; however, the elevated ALT returned to normal levels while the patient remained on fluoxetine. One patient transitioned from placebo to duloxetine (with abnormal ALT at study baseline) had a treatment-emergent ALT equal to or more than three times the upper limit of normal at the last study visit (week 36), which returned to near normal levels as the patient tapered off of duloxetine per protocol at completion of the study.
Safety: Suicidal ideation and behavior
Table 3 shows outcomes for suicidal ideation and behavior as classified using the C-SSRS. With the exception of one placebo-treated patient who reported suicidal ideation on the AE case report form, but not on the C-SSRS, all relevant events collected by spontaneous report on the AE case report forms were confirmed to be collected on the C-SSRS.
Patients are counted once in each category.
N=patients with baseline and ≥1 postbaseline observation; n=patients with event.
Lead-in baseline for acute treatment includes 2 screening visits (study baseline).
N=number of enrolled patients with ≥1 postbaseline suicidal ideation score and whose maximum C-SSRS suicidal ideation score during the lead-in baseline period is not missing and less than the maximum severity category.
N=number of enrolled patients whose suicidal ideation score is not missing and >0 during lead-in baseline.
N=number of enrolled patients without nonsuicidal self-injurious behavior at lead-in baseline visits and with a nonmissing postbaseline observation.
Lead-in baseline for extension treatment includes the last two visits of acute treatment.
Suicidal ideation: Wish to be dead, nonspecific active suicidal thoughts, active suicidal ideation with any methods (not plan) without intent to act, active suicidal ideation with some intent to act but without specific plan, and active suicidal ideation with specific plan and intent.
Suicidal behavior: Preparatory acts or behavior, aborted attempt, interrupted attempt, nonfatal suicide attempt, and completed suicide.
Suicidal acts: Nonfatal suicide attempt, and completed suicide.
There were no statistically significant differences between any treatment groups for any outcome on the C-SSRS during acute treatment. As assessed by the C-SSRS, suicidal ideation occurred in 14–15% of patients overall during acute treatment (that is, either preexisting or treatment-emergent); however compared with study baseline (lead-in), the suicidal ideation was treatment emergent (that is, new occurrence or any increase in severity from baseline) in fewer patients (7–8%) during acute treatment. During the extension treatment period, suicidal ideation occurred in 9–16% of patients overall; and compared with the end of acute treatment (lead-in), the suicidal ideation was treatment emergent in 9–4% of patients.
During the acute treatment period, one fluoxetine-treated patient (adolescent male) reported suicidal behavior (nonfatal suicide attempt), which was not considered an SAE. Upon follow-up with the site, the investigator reported, “The act was demonstrative without intent to die and without motive for harming self. There are no psychosocial stressors associated with the self-harm act.” As this follow-up was received after the acute database lock, the report of a nonfatal suicide attempt remains in the database.
During the extension treatment period, one patient (adolescent female) reported an SAE of suicidal ideation, which occurred ∼3 months after the patient had been transitioned from placebo to duloxetine. Also, during the extension treatment period, two patients (one duloxetine, one fluoxetine) experienced suicidal behavior (both nonfatal suicide attempts). The fluoxetine-treated patient (adolescent female) reported intentional overdose considered a suicide attempt and was discontinued because of this SAE. The duloxetine-treated patient (male child) was discontinued by his own decision (his mother did not believe medication was working) when the nonfatal suicide attempt non-SAE was reported; nonsuicidal self-injurious behavior (cutting) was also reported at the time of the nonfatal suicide attempt, and no other information was available for this patient.
Discussion
Efficacy results from this study indicated that duloxetine flexible dose (60–120 mg QD) was not statistically significantly different from placebo in the treatment of children and adolescents with MDD. In addition, the active control (fluoxetine) was not statistically significantly different from placebo on the primary outcome measure. Because neither the investigational drug (duloxetine), nor the active control (fluoxetine) separated from placebo on the primary outcome measure, the study is considered to be inconclusive.
Overall, the efficacy and safety results from this study were consistent with the sister study (Emslie et al. 2014) and the known TEAE and safety profile of duloxetine in adult patients (Cymbalta Full Prescribing Information). The only study design feature difference between this study and its sister study (Emslie et al. 2014) was that this study employed a flexible dosing schedule during acute treatment versus a fixed dosing during acute treatment in the sister study (Emslie et al. 2014). Similar to the previous open-label study (Prakash et al. 2012), the majority of patients (71%) in this study required dose escalation to higher duloxetine doses (90–120mg), and increased to those doses during acute treatment with overall less early discontinuation than in the fixed dosing sister study (Emslie et al. 2014). It is, therefore, interesting that the difference between drug and placebo on the primary efficacy measure at 10 weeks was less in this study than in the sister study (Emslie et al. 2014). The overall improvement of both duloxetine- and fluoxetine-treatment arms on the CDRS-R (24 points) in this study was generally consistent with fluoxetine improvement in previously published studies (Emslie et al. 1997, 2002), and the sister study (Emslie et al. 2014), suggesting that duloxetine dosing in this study and sister study (Emslie et al. 2014) was likely adequate, but that placebo response was high (discussion of which is included in Emslie et al. 2014). It is of note that these trials may point to the impact that frequency of visits (although minimized to the extent possible/acceptable according to treatment guidelines) and consistency of the nature of each appointment (assessment of efficacy and safety using the same measures at each study visit) has on pediatric depression, as the majority of children improved during the trials. The stability and support provided to the pediatric patients and their families while participating in the studies may have had a potentially therapeutic impact. Although there was a high response rate overall, the remission rate at 10 weeks was low (33–41%). However, remission rate increased substantially with continued care, and was >70% by 36 weeks. It is important for clinicians to encourage continued treatment beyond acute response, as the goal of treatment is remission of symptoms.
Although the design of this study and its sister study are quite similar, there were some notable differences in the implementation and study population characteristics. As with the sister study (Emslie et al. 2014), the treatment effect was consistent across all geographical regions in the study. Patients for this study, however, were primarily (∼60%) recruited in countries other than the United States versus ∼80% in the United States in the sister study (Emslie et al. 2014). This difference in the study implementation (geographic/cultural differences in the treatment of pediatric depression) may have led to some differences in the patient population, such as a greater proportion of patients (∼72%) experiencing a first episode of MDD in this study versus 42% in the sister study (Emslie et al. 2014). The lower proportion of patients with recurrent depression in this study is suggestive of a possibly less severely depressed patient population in this study, and may have had some impact on the smaller amount of separation between active drugs and placebo in this study (although baseline depression symptom severity was similar between the studies [CDRS-R total score of 58–60]). In addition, although rates of treatment-emergent suicidal ideation were similar between studies, there were a larger number of suicidal behaviors reported overall via C-SSRS in the sister study (eight cases) (Emslie et al. 2014) versus this study (three cases), which again suggests a possibly less severely depressed patient population in this study. It should be noted that with the exception of the one patient discussed in the study results, all events related to suicidal thoughts, behaviors, or nonsuicidal self-injurious behavior that were collected by spontaneous report on the AE case report forms during the 36 week study were confirmed to be collected on the C-SSRS; therefore, as is true for the sister study, the results of the C-SSRS provide the most complete information for this study.
Changes in weight were of particular interest given the pediatric patient population (expected to grow/gain weight over time). Regarding weight, results from this study and its sister study (Emslie et al. 2014) were consistent in that a statistically significant mean decrease in weight during acute treatment was observed for duloxetine- vs. placebo-treated patients who, on average, gained weight during acute treatment. However, also observed consistently in this study and its sister study (Emslie et al. 2014) was mean weight increase (recovery to baseline weight) over time during extension treatment. These results suggest that weight loss during acute treatment may be related to initial tolerability of duloxetine and are consistent with the pattern of weight change in duloxetine-treated adult patients (Cymbalta Full Prescribing Information). Elevations in blood pressure were expected based on the mechanism of action of duloxetine and results from the preliminary safety study of duloxetine in pediatric MDD patients (Prakash et al. 2012). Results from that study, however, were difficult to interpret, because of the absence of a placebo control (Prakash et al. 2012). Rates of PCS elevations in blood pressure at end-point for duloxetine during acute treatment were similar between this study (3–5%) and its sister study (2–5%) (Emslie et al. 2014), with no statistically significant differences between duloxetine and placebo in the incidence of PCS elevations of systolic or diastolic blood pressure at any time during acute treatment in either study. In this study, five duloxetine- and no fluoxetine-treated patients experienced sustained elevation in blood pressure over the 36 week study; however, in the sister study (Emslie et al. 2014), no duloxetine- and five fluoxetine-treated patients experienced sustained elevation in blood pressure over the 36 week study, which, given the different mechanisms of action for duloxetine and fluoxetine, suggests variability in blood pressure measurement. Finally, consistent with the sister study (Emslie et al. 2014) no new safety signals were identified based on laboratory or ECG results from this study.
Limitations
Several limitations must be taken into account when interpreting the results of this study and its sister study (Emslie et al. 2014). Patients with significant suicidal risk, comorbid psychiatric conditions requiring medication to manage, or other significant or unstable medical conditions, were excluded from this study. Therefore, the safety results from these studies have limited generalizability to a broader pediatric MDD population with greater suicide risk as well as greater psychiatric or medical burden. Also, because of the need to blind multiple doses of two different drugs, all patients were required to take six capsules of study drug per day. Although the capsule size was small (size 3, ∼4×14 mm), swallowing capsules may be difficult for pediatric patients, especially in the younger age range. Therefore, only a subset of pediatric patients who could swallow capsules was enrolled in this study. Finally, although the inclusion of active- and placebo-controlled arms helped in the interpretation of the study results, increasing the number of treatment arms, and thus the likelihood of receiving active drug versus placebo, may have also contributed to the placebo response observed in these studies.
Conclusions and Clinical Significance
Efficacy was not demonstrated in this study, as the results were inconclusive because neither the investigational drug (duloxetine) nor the active control (fluoxetine) demonstrated a statistically significant separation from placebo on the primary efficacy analysis of mean change from baseline to week 10 on the CDRS-R total score.
Overall, safety findings from this study were consistent with the known safety and tolerability profile for duloxetine.
Footnotes
Acknowledgments
The authors thank the principal investigators and their clinical staff. The authors also thank the many patients and their families who generously agreed to participate in this clinical trial, and the clinical operations staff and statistical analysts of the Duloxetine Antidepressant Team for their excellent implementation of the trial.
Disclosures
Sarah D. Atkinson has research grant affiliations with Amgen Pharmaceuticals; AstraZeneca; Bristol Myers Squibb; Cephalon (Teva); ENDO Pharmaceuticals; Grunenethal (past only); Johnson & Johnson (past only); Lundbeck; Merck, Sharpe, Dome; Naurex; Novartis (past only); Otsuka; Purdue (past only); Roche Genentech; Sanofiaventis; Sunovion; Targacept; and VANDA.
Graham J. Emslie has received research support from BioMarin, Duke University, Eli Lilly and Company, Forest Laboratories, GlaxoSmithKline, and Mylan, and has served as a consultant for Allergan, BioBehavioral Diagnostics Inc., Eli Lilly and Company, INC Research Inc., Lundbeck, Pfizer, Seaside Therapeutics, Shire, the Texas Department of State Health Services, Valeant, and Wyeth. Now an emeritus faculty member, John S. March has no established relationships with the pharmaceutical industry. Dr. March has served as a consultant or scientific advisor to Attention Therapeutics, Bristol Myers Squibb, Eli Lilly and Company, and Pfizer; received study drug for a National Institute of Mental Health (NIMH)-funded study from Eli Lilly and Company and Pfizer; is an equity holder in MedAvante; and receives royalties from Guilford Press, Oxford University Press, and Multihealth Systems. Dr. March has received research support from National Institute on Drug Abuse (NIDA), NIMH, and Pfizer. Dr. March has not engaged in promotional work; for example, being part of a speakers bureau or training, for >15 years. Apurva Prakash, Qi Zhang, Beth A. Pangallo, and Mark E. Bangs are employees of and own stock or equity in Eli Lilly and Company.