
Abstract
Objective:
Disruptive or challenging behavior problems pose a threat to children and adolescents with intellectual disabilities and their caregivers. Psychopharmacological treatment is mostly studied with new-generation antipsychotics and has been criticized for adverse side effects. This study examined the effect of the classic antipsychotic zuclopenthixol.
Methods:
A total of 39 boys (ages 8.0–17.11 years) with learning disabilities were included and examined for a response to zuclopenthixol during a 6 week period of open label treatment. Doses started low and were adapted individually. From responders, zuclopenthixol was randomly withdrawn for 12 weeks. Responses to withdrawal were observed by external raters using the Modified Overt Aggression Scale.
Results:
Of all patients included into the study, 15 were not randomized because of insufficient therapeutic effect, adverse event, or noncompliance. Kaplan–Meier estimations showed less aggressive behavior problems for the continuing subgroup (n=9) than in the placebo group (n=15). Individual doses stayed <10 mg/day.
Conclusions:
Zuclopenthixol proved to be effective in reducing challenging behavior in boys even at low doses.
Introduction
C
Recent 6 week randomized, double-blind, placebo-controlled studies of antipsychotic drugs focusing on risperidone revealed good effects on aggression and self-injurious behavior in children and adolescents with ID (Buitelaar et al. 2001, Snyder et al. 2002). Risperidone however, produces adverse effects such as weight gain, somnolence, headache, mild to moderate extrapyramidal motor symptoms (EPMS), and elevated serum prolactin in children (Handen and Gilchrist 2006). The question remained whether classic antipsychotics given in small doses could effectively reduce challenging behavior problems, while keeping the burden of adverse side effects low in children and adolescents with ID. The classic antipsychotic zuclopenthixol was successfully used to treat challenging behavior among adults (Haessler et al. 2007). Zuclopenthixol, a polycyclic thioxantene (C22H25ClN2OS), works as a dopamine and serotonin receptor antagonist. Working on two receptor types makes zuclopenthixol an antipsychotic drug with fewer side effects than other classic antipsychotics. It was approved for almost all European countries, China, Canada, Australia, countries from South and Middle America, Israel, and South Africa. In the United States, zuclopenthixol is not approved. Here, the thioxantene derivate Navane®, a thiothixine, is approved for the treatment of schizophrenia, but not for challenging behavior.
To our knowledge, the study reported here is the first multicenter, double-blind, placebo-controlled trial of zuclopenthixol involving children and adolescents with ID displaying aggressive behavior problems.
Methods
A randomized, double-blind, placebo-controlled withdrawal study for parallel groups was conducted in two German centers. After approval from the Ethics Committee, the study was conducted from January 2009 to June 2013 in compliance with the Declaration of Helsinki and all applicable legal requirements in Germany (EudraCT number 2008-000584042). Thirty-nine male patients ages 8–17.11 years showing moderate to low ID (intelligence quotient [IQ] 30–84) received open treatment with zuclopenthixol for 6 weeks, because of aggressive behavior. Eligible patients were children and adolescents admitted to psychiatric hospitals for challenging behavior rated below the critical value of 39 for complex VII “Aggression” of the Disability Assessment Schedule (Holmes et al. 1982). After a complete description of the study was given to the patients and their legal representatives, voluntary written informed consent or assent was obtained from both the patients and their legal guardians for participation and publication of study results. After open label treatment, responders (n=24) were randomized to continue or discontinue treatment for up to 12 weeks. Discontinued patients received placebo medication after randomization. The null hypothesis was that the two groups would not differ in their recurrence rates after withdrawal of the medication.
For all patients the Modified Overt Aggression Scale (MOAS) (Yudofsky et al. 1986) was administered every 2 weeks during the entire trial. Several secondary measures, medical history, and safety measures, including possible withdrawal symptoms, extrapyramidal signs, vital signs, and weight were recorded. Routine laboratory tests of prolactin and serum levels of zuclopenthixol were conducted.
The primary efficacy measures were binary variables derived from weighed sums of the MOAS aggression scores. The weighing of the MOAS aggression scores has a higher impact on severe (physical) forms of aggression (Kay et al. 1988). Patients with an aggravation of aggressive behavior indicated by a deterioration of≥3 points in MOAS sum scores at two subsequent visits when compared with their state either at first visit (open period) or at randomization (withdrawal period) were called “nonresponders,” and left the study. All patients without deterioration were interpreted as “responders” unless they left the study because of disallowed concomitant treatment, adverse events, or other reasons violating the protocol.
During the open treatment period, zuclopenthixol was administered at a dose of 4–20 mg/day. The dose was adjusted once or twice daily as judged necessary by the clinician. After randomization the individual dose was kept as stable as possible. The mean of daily doses given throughout the whole trial was 7.90 mg/day (standard deviation=2.61 mg/day). Concomitant use of other antipsychotics was forbidden throughout the study. The use of constant doses of anticonvulsants (different types), lithium, antiextrapyramidal-symptom medication, and benzodiazepines as an antiepileptic escape medication was permitted if the dose had been stable for at least 30 days before the start of the study.
In cases of extremely aggravated agitation, escape medications were allowed. All concomitant medications were recorded.
Exclusion criteria were the following: diagnosed neurological disorders (excluding epilepsy), psychotic disorders, infantile cerebral palsy, hypersensitivity to zuclopenthixol, and cardiac abnormalities.
Results
The proportion of responders during the open period was 62% for the sample to be treated (24/39). Approximately 18% of all participants (7/39) were excluded during the open label period because of adverse effects (see Fig. 1). All adverse side effects were regarded as EPMS, including glossopharyngeal spasms. EPMS were treated for 2 days with biperiden. In case in which EPMS could not be reduced to a tolerable level, the primary medication was terminated, and the subjects left the study. Drowsiness, not regarded as an adverse side effect, was reported for ∼50% of boys during the open period. Six participants did not respond to the medication during the open label period and had to be excluded from randomization. After randomization, three persons had to be excluded for other reasons or for violating the protocol. The proportion of responders among participants remaining in the study for the entire withdrawal period was significantly larger in the zuclopenthixol group (6/7 86%), than in the placebo group (4/14, 29%) (difference: 57%, χ2=6109, df=1, p value from Fisher's exact test: 0.024).
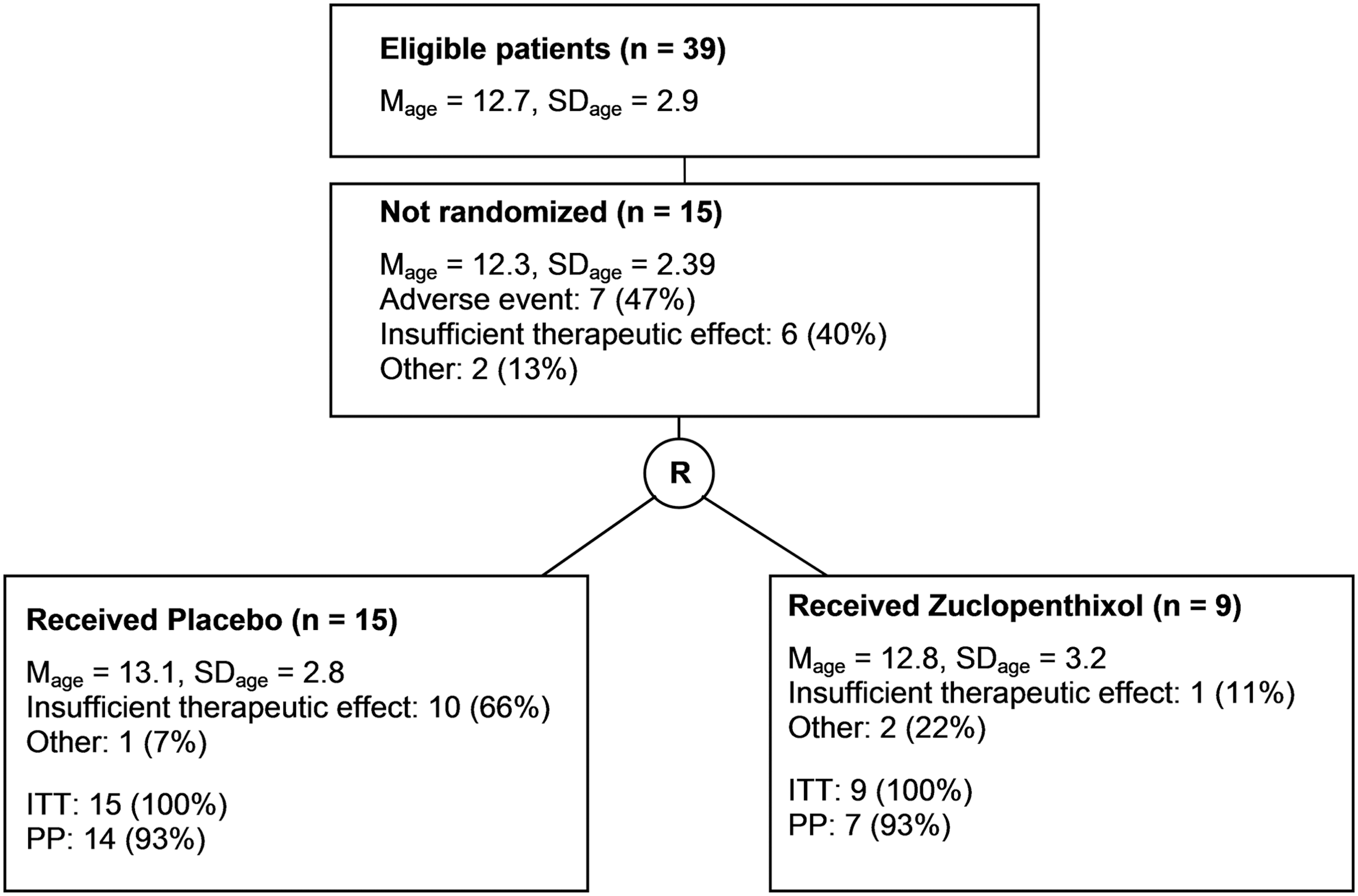
Study flow for the open and withdrawal period (M=mean, SD=standard deviation, ITT=sample intended to be treated, PP=sample per protocol).
Figure 2 shows the Kaplan–Meier estimates of responder rates for the placebo group and for the zuclopenthixol group during the withdrawal period. Following the results of a log-rank test performed on both survival functions the null hypothesis was rejected (p value: 0.024).
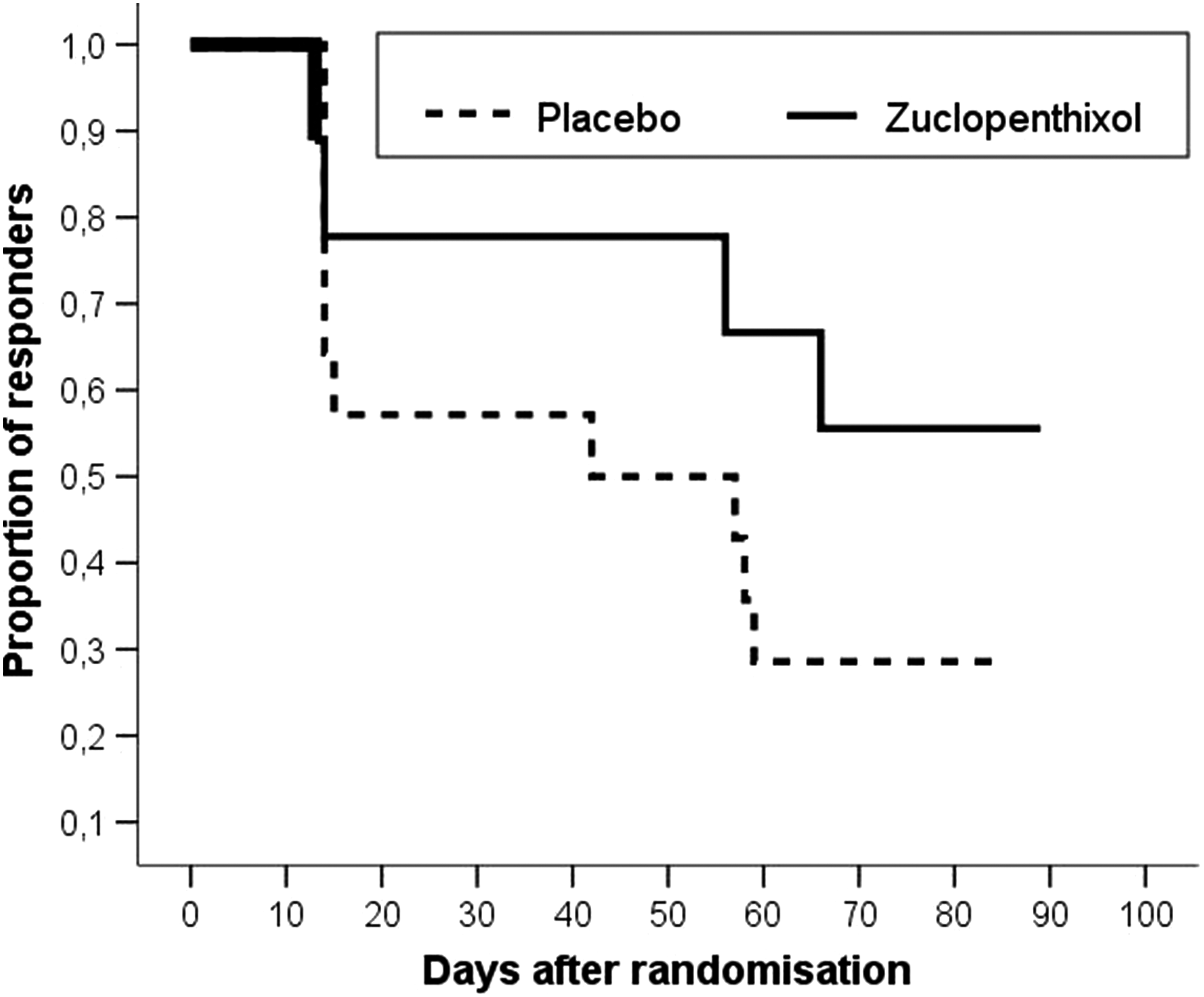
Kaplan–Meier estimates of responder rates after randomized withdrawal of zuclopenthixol based on the weighed sum of scores from the Modified Overt Aggression Scale.
Psychotropic comedications during the withdrawal period appeared to be equally distributed across the groups regarding anticholinergic and antiepileptic medication (verum=1/9, 11.11%; placebo=2/15, 13%). Possible symptoms of withdrawal, such as nausea, insomnia, or diarrhea were recorded and did not differ between the groups. No adverse side effects were recorded for the time after randomization. The number of reports about drowsiness did not differ between groups after randomization.
Discussion
As was shown for adults (Haessler et al. 2007), this discontinuation study found zuclopenthixol to be significantly superior to placebo in reducing aggressive behavior in children and adolescents with ID. The positive effect of the typical antipsychotic zuclopenthixol occurred even at a low average dosage of 7.90 mg per day. EPMS were twice as likely to occur as side effects, compared with in adults (8%) with ID (Haessler et al. 2008) and were displayed by approximately one out of five boys. Compared with a study on risperidone (Buitelaar et al. 2001) conducted with adolescents, we found EPMS to be somewhat less dominant. In this Dutch study frequencies up to 26% of mild EPMS for the verum group at the end-point of study were reported. In the study presented here, EPMS occurred for 18% of the sample to be treated, including during the open period. Limitations of this study were the exclusion of girls, the high rate of dropouts during the open label period (38%), and a small sample size.
Conclusions
For boys tolerating and responding to zuclopenthixol, it proved to be effective at low doses 4 months after applying it for the first time. Adverse side effects occur more often than in adults, limiting the tolerability among children and adolescents.
Clinical Significance
The classic antipsychotic zuclopenthixol provides a safe and cost-effective alternative to new-generation antipsychotics for the treatment of challenging behavior not only in adults, but also in boys. It can be given at low dosages in order to minimize adverse side effects. Promising responses, but also unwanted side effects, can be monitored during the first 6 weeks. Early responses to the drug should be monitored carefully.
Footnotes
Disclosures
Frank Hässler is director of an institution that received financial support from Bayer Vital GmbH to conduct the study as an investigator-initiated trial. He worked at Advisory Boards for Eli Lilly GmbH Germany, Janssen-Cilag, and Shire, and received research support from Novartis Pharmaceuticals, and Bayer Vital. He received travel grants from AstraZeneca Pharmaceutical, Bayer Vital, and Novartis Pharmaceuticals. He also received consulting fees from Bayer Vital, Janssen-Cilag, and Novartis Pharmaceuticals. Alexander Dück and Olaf Reis work at an institution that received a grant from Bayer Vital GmbH to conduct the study. Martin Jung has no conflicts of interest.