
Abstract
Objective:
To assess acute and longer-term safety of duloxetine in the treatment of children and adolescents with major depressive disorder (MDD), a pooled analysis of data from two completed randomized, double-blind, multicenter, phase 3, placebo- and active-controlled trials was undertaken. In these studies, neither duloxetine (investigational drug) nor fluoxetine (active control) demonstrated a statistically significant improvement compared with placebo on the primary efficacy measure.
Methods:
Patients ages 7–17 years with MDD as defined by the Diagnostic and Statistical Manual of Mental Disorders, 4th ed., Text Revision (DSM-IV-TR) received duloxetine (n=341), fluoxetine (n=234), or placebo (n=225) for 10 week acute and 26 week extended (duloxetine or fluoxetine only) treatments. Safety measures included treatment-emergent adverse events (TEAEs), the Columbia-Suicide Severity Rating Scale, vital signs, electrocardiograms, laboratory samples, and growth (height and weight) assessments.
Results:
Significantly more patients discontinued because of adverse events during duloxetine (8.2%) treatment than during placebo (3.1%) treatment (p≤0.05). TEAEs in >10% of duloxetine-treated patients were headache and nausea. No completed suicides or deaths occurred. During acute treatment, 6.6% of duloxetine-, 8.0% of fluoxetine-, and 8.2% of placebo-treated patients had worsening suicidal ideation from baseline. Among patients initially randomized to duloxetine or fluoxetine who had suicidal ideation at study baseline, 81% of duloxetine- and 77% of fluoxetine-treated patients had improvements in suicidal ideation at end-point in the 36-week studies. Suicidal behavior occurred in two fluoxetine-treated patients and one placebo-treated patient during acute treatment, and in seven duloxetine-treated patients and one fluoxetine-treated patient during extended treatment. Duloxetine-treated patients had a mean pulse increase of ∼3 beats per minute, and mean blood pressure (both systolic and diastolic) increases of <2.0 mm Hg at week 36. Weight decrease (≥3.5%) during acute treatment occurred with statistically (p≤0.05) greater frequency for both the duloxetine (11.4%) and fluoxetine (11.5%) groups versus the placebo (5.5%) group; however, mean weight increase occurred for both duloxetine and fluoxetine groups during extended treatment.
Conclusion:
Results from this pooled analysis of two studies were consistent with the known safety and tolerability profile of duloxetine.
Clinical Trial Registry Numbers:
NCT00849901 and NCT00849693.
Introduction
M
Large randomized controlled trials (RCTs) play an important role in characterizing the safety and tolerability profile of new drugs and informing regulatory decisions whether to authorize the use of new drugs. Among studies of antidepressants in the pediatric MDD population, SSRIs have been the most commonly studied class of antidepressants, with fewer studies of antidepressants with other mechanisms of action, such as mirtazapine or venlafaxine (Bridge et al. 2009). Duloxetine is a selective serotonin and norepinephrine reuptake inhibitor (SNRI) and has been approved for the treatment of MDD in adults (Cymbalta prescribing information). Two randomized, placebo-controlled studies were conducted to investigate the efficacy and safety of duloxetine for the treatment of children and adolescents with MDD (Atkinson et al. 2014; Emslie et al. 2014). In both studies, fluoxetine was included as an active pharmacological control with a known efficacy and safety profile in the pediatric MDD patient population.
Duloxetine dosing in the two pediatric MDD RCTs was based on a previous safety, tolerability, and pharmacokinetic study, the results of which suggested that adjustment of total daily dose based on body weight or age is not warranted for pediatric patients, and that different total daily doses may not be warranted for pediatric patients relative to adults (Prakash et al. 2012). Therefore, the full range of doses approved for use in the adult population was used in the duloxetine pediatric MDD RCTs. In these two pediatric MDD RCTs, neither the investigational drug (duloxetine) nor the active control (fluoxetine) demonstrated a statistically significant separation from placebo on the primary efficacy analysis, thus rendering the studies inconclusive (Atkinson et al. 2014; Emslie et al. 2014). However, mean improvements on the Children's Depression Rating Scale-Revised (∼22 points) and response rates (>50%) in duloxetine- and fluoxetine-treated patients from the duloxetine pediatric MDD RCTs were consistent with historical improvements in fluoxetine-treated patients from positive studies (Emslie et al. 1997, 2002).
The duloxetine pediatric MDD RCTs were unique because they were the first studies in pediatric MDD patients to include both a placebo and an active control (fluoxetine) with established efficacy and safety in the pediatric MDD patient population. The design of the two duloxetine pediatric MDD RCTs allowed for comparisons of duloxetine (an SNRI) to placebo and fluoxetine (an SSRI) during a 10 week acute treatment phase and between duloxetine and fluoxetine over the full 36 week longitudinal course of the studies. The studies also incorporated the Columbia Suicide Severity Rating Scale to allow for the prospective assessment of suicidal ideation, behavior, and nonsuicidal self-injurious behavior at all visits over the entire 36 week length of the studies. The primary efficacy and safety results for the two duloxetine pediatric MDD RCTs have been previously disclosed (Atkinson et al. 2014; Emslie et al. 2014); however, given the similarities in design between the two studies, we completed a pooled analysis of the studies for the purpose of assessing safety outcomes using the larger sample size (n=800). Specifically, this pooled analysis includes a summary of results of treatment-emergent adverse events (TEAEs), serious adverse events (SAEs), discontinuations due to adverse events (AEs), as well as the Columbia-Suicide Severity Rating Scale (C-SSRS), cardiovascular outcomes, laboratory outcomes, and growth outcomes not addressed in the original disclosures.
Methods
Study design
The current report is a pooled analysis of safety data from two randomized, double-blind, multicenter, Phase 3, placebo- and active-controlled clinical trials. Each trial was approved by the ethics review boards for each study site and conducted in accordance with good clinical practice guidelines. In accordance with the principles of the Declaration of Helsinki, a parent/legal representative of each study patient provided written informed consent prior to administration of any study drug or study procedures, and patients (children and adolescents) also provided assent to participate in the study as appropriate. Patients participated in the studies at 125 psychiatric clinical sites in 12 countries (Argentina, Canada, Estonia, Finland, France, Germany, Mexico, Russia, Slovakia, South Africa, Ukraine, and the United States) from March 2009 to October 2011.
The primary objective of each of the two studies included in this pooled analysis was to assess the efficacy of duloxetine in the treatment of children and adolescents with MDD.
Each study included a 10 week placebo- and fluoxetine-controlled acute treatment phase, a 26 week double-blind fluoxetine-controlled extension treatment phase, and a 2 week double-blind drug tapering phase. During the acute treatment phase, patients received placebo, active drug fixed doses (duloxetine 30 or 60 mg once daily [q.d.] or fluoxetine 20 mg q.d., Emslie et al. 2014), or active drug flexible doses (duloxetine 60–120 mg q.d. or fluoxetine 20–40 mg q.d., Atkinson et al. 2014).
A 26 week, double-blind extended treatment phase followed acute treatment in each of the two studies. During extended treatment in both studies, patients who were initially randomized to duloxetine or fluoxetine continued on a flexible-dose regimen of that drug (duloxetine 60–120 mg q.d. or fluoxetine 20–40 mg q.d.), and patients who were initially randomized to placebo were transitioned to duloxetine (flexible dosing) after the 10 week time point. Patients discontinuing from the studies (early termination or 36 week completers) entered a two week, double-blind, drug-tapering phase based on investigators' determination of patient safety. Patients receiving duloxetine 90 or 120 mg q.d. at the last study visit had their doses reduced to 60 mg q.d. for the1st week of the tapering phase and to 30 mg q.d. for the 2nd week; patients receiving duloxetine 60 mg q.d. at the last study visit had their doses reduced to 30 mg q.d. for the 1st week of the tapering phase and received placebo during the 2nd week; and those patients receiving duloxetine 30 mg q.d. at the last study visit received placebo for both weeks of the tapering phase. Patients receiving fluoxetine 40 mg q.d. at the last study visit had their doses reduced to 20 mg q.d. for the 1st week of the tapering phase and 10 mg q.d. for the 2nd week; patients receiving fluoxetine 20 mg q.d. at the last study visit had their doses reduced to 10 mg q.d. for the 1st week of the tapering phase and received placebo during the 2nd week. Patients receiving placebo in the acute phase received placebo for both weeks of the tapering phase.
Assessments
Scheduled study visits occurred at weeks 1, 2, 4, 7, and 10 during the acute treatment period and subsequently every 2–4 weeks until week 36 during the extension treatment period. AEs were collected by spontaneous report at all visits. Suicidal ideation and behavior and non-suicidal self-injurious behavior information was solicited using the Columbia Suicide Severity Rating Scale (C-SSRS) at every visit, and any suicidal ideation or behavior or self-injurious events that were reported spontaneously during AE collection were confirmed to be reported on the C-SSRS (with the exception of 1 case described in Atkinson et al. 2014). Blood pressure (BP) and pulse were collected at every visit. Electrocardiograms were collected at baseline and weeks 10, 24, 36, or early discontinuation. Laboratory samples were collected at baseline and weeks 4, 10, 14, 20, 24, 36, or early discontinuation. Weight was collected at every visit, and height was collected at baseline and weeks 10, 24, 38, or early discontinuation.
Patients
Study participants were outpatient children (7–11 years) and adolescents (12–17 years) who met Diagnostic and Statistical Manual of Mental Disorders, 4th ed., Text Revision (DSM-IV-TR) criteria for MDD without psychotic features. Patients were excluded from the studies if they were pregnant or lactating or had baseline weights <20 kg; current Axis I disorders (other than MDD) requiring pharmacotherapy; histories of bipolar disorder, schizophrenia, or any other psychotic disorder; one or more first degree relatives with bipolar I disorder; serious or unstable medical illnesses; serious suicide risk; histories of substance abuse or dependence within the last year; or unexplained positive urine drug screening for substances of abuse.
Statistical methods
Analyses were conducted on an intent-to-treat population. All tests of hypotheses were based on the significance level of 0.05. No adjustments for multiple comparisons were made. Analyses of continuous safety variables were performed using a mixed model for repeated measures (MMRM), an analysis of variance (ANOVA) model, or an analysis of covariance (ANCOVA) model with a last-observation-carried-forward (LOCF) end-point. The ANOVA model contained the main effects of treatment and study. The ANCOVA model was the ANOVA model with baseline added as a covariate. “Mean change” refers to adjusted least square (LS) mean change from the MMRM, ANOVA, or ANCOVA model.
For the MMRM analysis, the model included the fixed categorical effects of treatment, study, visit, treatment-by-visit interaction, age category (children, 7–11 years; adolescents, 12–17 years), and age category-by-visit interaction, as well as the continuous, fixed covariates of the baseline value being analyzed and baseline value of the variable being analyzed in the analyzed-by-visit interaction. For categorical data, the significance of overall treatment-group differences were assessed primarily using the Cochran–Mantel–Haenszel (CMH) general association test adjusting for the effect of study. For extension phase-specific analyses, no statistical comparisons were made between treatment groups (patients initially randomized to duloxetine or fluoxetine and patients who were transitioned from placebo to duloxetine) because of potential selection bias. For longitudinal analyses over the combined acute and extension phases, statistical comparisons between treatment groups (patients initially randomized to duloxetine and those initially randomized to fluoxetine) were made.
For growth analyses of height and weight, a normalized score (z score) was calculated based on age- and sex-matched reference data from the Centers for Disease Control (National Center for Health Statistics 2000). For a subgroup analysis of z scores, an ANCOVA model with terms for study, treatment, baseline z score, age category, subgroup, and subgroup-by-treatment interaction was used to assess the subgroup and subgroup-by-treatment interaction effects. Comparisons between treatment groups within subgroups were made using an ANCOVA model with terms for baseline z score, study, treatment, and age category.
Potentially clinically significant (PCS) changes in BP, pulse, weight, and electrocardiogram values were defined as follows.
PCS high BP: Increase of ≥5 mm Hg from highest baseline BP to a value above the 95th percentile based on age, height, and sex (National High Blood Pressure Education Program Working Group on High Blood Pressure in Children and Adolescents 2004)
Sustained elevation in BP: PCS high BP at three consecutive postbaseline visits
PCS low systolic BP: Decrease of ≥15 mm Hg from baseline low to a value ≤80 mm Hg for children and decrease of ≥20 mm Hg from baseline low to a value ≤90 mm Hg for adolescents
PCS low diastolic BP: Decrease of ≥10 mm Hg from baseline low to a value ≤50 mm Hg
PCS high pulse: Increase of ≥15 beats per minute (bpm) from baseline high to a value >140 bpm for children or an increase of ≥15 bpm from baseline high to a value >120 bpm for adolescents
PCS low pulse: Decrease of ≥25 bpm from baseline low to a value of <60 bpm for children or a decrease of ≥15 bpm from baseline low to a value <50 bpm for adolescents
PCS weight loss: decrease ≥3.5% from baseline low
Abnormalities in electrocardiogram values were defined as follows.
Heart rate abnormal high: >130 bpm for children or >110 bpm for adolescents
Heart rate abnormal low: <60 bpm for children or <50 bpm for adolescents
PR interval high: >220 msec
QRS interval high: >120 msec
QTcF high: ≥470 msec for female patients or ≥450 msec for male patients
QTcF abnormal increase: >40 msec increase from baseline
QTcF PCS high: >500 msec
Definitions of treatment- and discontinuation-emergent events, suicidal ideation, and suicidal behavior were as follows.
TEAEs or treatment-emergent suicidal ideation or behavior during the acute treatment phase: new event or worsening of event (any increase in severity category) compared with the study screening period
TEAEs or treatment-emergent suicidal ideation or behavior during the extension treatment phase: new event or worsening of event (any increase in severity category) compared with the last two visits of the acute treatment phase
Treatment-emergent suicidal ideation or behavior over the 36 week course of the study (combined acute and extension phases): new event or worsening of event (any increase in severity category) compared with the study screening period
Discontinuation-emergent AE or discontinuation-emergent suicidal ideation or behavior during the drug tapering phase: new event or worsening of event (any increase in severity category) compared with the last two visits of the treatment phase before entering the tapering phase
Results
Patients
The mean age of randomized patients was 13.1 years (median 13.3 years) with 51.6% female and 48.4% male, 40.9% children (7–11 years), and 59.1% adolescents (12–17 years).
TEAEs during acute treatment
There were no statistically significant differences between the active drug groups or between either active drug group and the placebo group in the proportion of patients reporting TEAEs within the subgroups of children (7–11 years) or adolescents (12–17 years). During the 10 week acute treatment phase, TEAEs were reported in 83/140 (59.3%) duloxetine-, 61/100 (61.0%) fluoxetine-, and 56/87 (64.4%) placebo-treated children; and in 133/201 (66.2%) duloxetine-, 84/134 (62.7%) fluoxetine-, and 83/138 (60.1%) placebo-treated adolescents. Headache and gastrointestinal events were the most commonly reported TEAEs in duloxetine-treated patients during acute treatment. TEAEs reported with statistically significantly greater incidence for the duloxetine group than for the placebo group were nausea, diarrhea, and sedation; whereas nausea and dizziness were reported with statistically significantly greater incidence for the duloxetine group than for the fluoxetine group (Table 1). Abnormal dreams were reported with statistically significantly greater incidence for the fluoxetine group than for the placebo group (Table 1).
TEAEs reported with frequency≥5% in either the duloxetine or fluoxetine group or those reported with statistically significantly (p≤.05) greater frequency for either active drug group (duloxetine or fluoxetine) compared with the placebo group during the acute treatment phase.
Statistical comparisons between treatment groups were not conducted for the extension phase analyses because of selection bias (lack of randomization).
TEAEs reported with frequency≥5% in any treatment group during the extension treatment phase.
p≤.05 vs. placebo.
p≤.05 duloxetine vs. fluoxetine.
TEAE, treatment-emergent adverse events.
SAEs during acute treatment
During the acute treatment phase, a total of eight duloxetine-treated patients experienced 10 SAEs, eight fluoxetine-treated patients experienced 11 SAEs, and three placebo-treated patients experienced 4 SAEs (described in Atkinson et al. 2014 and Emslie et al. 2014), with no statistically significant differences reported between active drug groups (duloxetine or fluoxetine) and the placebo group. For most patients who experienced more than one SAE during acute treatment, the SAEs occurred at approximately the same time and resulted in hospitalization for the multiple SAEs (Table 2).
All SAEs reported during the acute treatment phase are shown.
All SAEs reported during the extension treatment phase are shown.
Statistical comparisons between treatment groups were not conducted for the extension phase analyses because of selection bias (lack of randomization).
Not considered a suicide attempt as the overdose was taken without intent to die, per investigator.
Self-injurious behavior and hallucination (2 SAEs in 1 patient, same hospitalization).
Drug abuse and panic attack (2 SAEs in 1 patient, same hospitalization).
Suicide attempt and depression (2 SAEs in 1 patient, same hospitalization).
Suicide attempt and wound (2 SAEs in 1 patient, same hospitalization).
Irritable bowel syndrome in one patient (preexisting condition in acute phase which worsened to SAE in the extension phase).
Suicide attempt.
Suicidal ideation and aggression (2 SAEs in 1 patient, same hospitalization).
Aggression and abnormal behavior (2 SAEs in 1 patient, same hospitalization).
Gastritis and lymphadenitis (2 SAEs in 1 patient, 2 separate hospitalizations).
Somnolence in one patient (SAE in acute phase which resolved during the extension phase).
Suicide attempt by intentional overdose (2 SAEs in 1 patient, same hospitalization).
Epilepsy and convulsion (2 SAEs in 1 patient, same hospitalization).
Suicide attempt and homicidal ideation (2 SAEs in 1 patient, same hospitalization).
Major depression in one patient (SAE during acute treatment which resolved during extension treatment)
Suicidal ideation and restlessness (2 SAEs in 1 patient, same hospitalization).
p≤0.05 duloxetine vs. fluoxetine.
SAE, serious adverse event.
Discontinuations because of AEs during acute treatment
A statistically significantly greater proportion of duloxetine-treated patients discontinued because of an AE compared with both fluoxetine- and placebo-treated patients (Table 3). Among the 19 patients experiencing SAEs during acute treatment, 12 were discontinued from the studies because of the SAE (7 duloxetine, 4 fluoxetine, and 1 placebo). The majority of SAEs and AEs leading to discontinuation were gastrointestinal and psychiatric events (Tables 2 and 3).
Statistical comparisons between treatment groups were not conducted for the extension phase analyses because of selection bias (lack of randomization).
Serious adverse event (SAE)
SAE in one of the two reported cases in the duloxetine group.
p≤.05 duloxetine versus placebo.
p≤.05 duloxetine versus fluoxetine.
TEAEs, SAEs, and discontinuations because of AEs during extended treatment
During the 26 week extended treatment phase, similar to acute treatment, gastrointestinal events were again the most frequently reported TEAEs, and the majority of SAEs and AEs leading to discontinuation were gastrointestinal and psychiatric events (Tables 1 –3). During the extension treatment phase, a total of six duloxetine-treated patients experienced eight SAEs, four fluoxetine-treated patients experienced six SAEs, and seven duloxetine-treated patients transitioned from placebo experienced eight SAEs (described in Atkinson et al. 2014 and Emslie et al. 2014). For most patients who experienced more than one SAE during extension treatment, the SAEs occurred at approximately the same time and resulted in hospitalization for the multiple SAEs (Table 2). Among the 17 patients experiencing SAEs during extension treatment, 8 were discontinued from the studies because of the SAE (5 duloxetine and 3 fluoxetine).
Discontinuation-emergent AEs
During the 2 week drug tapering phase, discontinuation-emergent AEs reported by more than one patient while discontinuing duloxetine were headache, dizziness, insomnia/initial insomnia, paraesthesia, upper abdominal pain, irritability, nausea, depression, and seasonal allergy; whereas only headache was reported in more than one patient discontinuing from fluoxetine. During the tapering phase, one SAE (accidental fracture of thoracic vertebra) was experienced by a patient discontinuing fluoxetine.
Suicidal ideation and behavior results from the C-SSRS
No completed suicides or deaths occurred in these studies. Rates of treatment-emergent suicidal ideation, suicidal behaviors, or nonsuicidal self-injurious behavior during acute treatment and separately for extension treatment are shown in Table 4.
Patients are counted once within each category, but may be counted in multiple different categories.
Lead-in baseline for the acute treatment period includes the two screening visits (study baseline). Treatment-emergent is defined as any new event or any worsening in severity of the event from the lead-in baseline.
N, number of enrolled patients with at least one postbaseline suicidal ideation score and whose maximum C-SSRS suicidal ideation score during the lead-in baseline period is not missing and less than the maximum severity (category 5).
N, number of enrolled patients whose suicidal ideation score is not missing and greater than 0 during lead-in baseline.
N, number of enrolled patients without non suicidal self-injurious behavior at lead-in baseline visits and with nonmissing postbaseline.
Lead-in baseline for the extension treatment period includes the last two visits of the acute treatment period. Treatment-emergent is defined as any new event or any worsening in severity of the event from the lead-in baseline.
p≤.05 versus placebo.
Suicidal ideation includes wish to be dead, nonspecific active suicidal thoughts, active suicidal ideation with any methods (not plan) without intent to act, active suicidal ideation with some intent to act, without specific plan, and active suicidal ideation with specific plan and intent; suicidal behavior includes preparatory acts or behavior, aborted attempt, interrupted attempt, nonfatal suicide attempt, and completed suicide; nonsuicidal self-injurious behavior includes intentional self-injury without intent to commit suicide.
N, patients with baseline and at least one postbaseline observation; n, patients with an event.
Over the 36 week course of the studies (acute plus extension phases), for patients initially randomized to duloxetine and those initially randomized to fluoxetine, there were no statistically significant differences on any suicide-related outcome (ideation or behavior) or nonsuicidal self-injurious behavior between groups. Suicidal behavior was reported in 6/333 (1.8%) patients initially randomized to duloxetine and was reported in 3/225 (1.3%) patients initially randomized to fluoxetine. Treatment-emergent suicidal ideation (i.e., any new ideation or any worsening in severity of ideation from study baseline screening period) during the 36 weeks of treatment was reported in 37/333 (11.1%) patients initially randomized to duloxetine and in 34/225 (15.1%) patients initially randomized to fluoxetine. Among patients with suicidal ideation at study baseline, improvement in suicidal ideation (reduction in severity from baseline to end-point) during the 36 weeks of treatment was observed in 42/52 (80.8%) patients initially randomized to duloxetine and in 27/35 (77.1%) patients initially randomized to fluoxetine. Treatment-emergent nonsuicidal self-injurious behavior (any newly reported after study baseline screening period) during the 36 weeks of treatment was reported in 18/328 (5.5%) patients initially randomized to duloxetine and in 10/224 (4.5%) patients initially randomized to fluoxetine.
During the 2 week drug tapering phase, discontinuation-emergent suicidal ideation (new ideation or any worsening in severity of ideation from the end of the treatment phase) was reported in 3/276 (1.1%) patients discontinuing duloxetine and in 2/124 (1.6%) patients discontinuing fluoxetine. There were no reports of nonsuicidal self-injurious behavior or suicidal behavior in any patients discontinuing duloxetine or fluoxetine treatment.
Cardiovascular data
Mean baseline systolic BP, diastolic BP, and pulse rate were ∼109 mm Hg, 68 mm Hg, and 77 bpm, respectively. There were no statistically significant differences between either of the active drug groups (duloxetine or fluoxetine) and the placebo group in mean change from baseline to 10 weeks in sitting BP or pulse; however, a statistically significant difference was observed between duloxetine and fluoxetine in sitting pulse (Table 5). During the acute treatment phase, there were no statistically significant differences between treatment groups in the incidence of PCS BP or pulse rate, and there were no statistically significant differences between treatment groups in the incidence of abnormal or PCS electrocardiogram values (Table 5).
Baseline was defined as last observation during the screening period, and end-point was the 10 week end-point of the acute treatment phase (mixed model repeated measures [MMRM]).
N, number of patients with low or normal values at baseline and with at least one non-missing postbaseline measure; n, number of patients with a PCS postbaseline measurement.
N, number of patients with high or normal values at baseline and with at least one non-missing postbaseline measure; n, number of patients with a PCS postbaseline measurement.
N, number of patients with normal baseline value and at least one postbaseline measurement; n, number of patients with abnormal or PCS observation
*p≤.05 versus placebo.
p ≤.05 duloxetine versus fluoxetine.
PCS high blood pressure (BP), increase ≥5 mm Hg from highest baseline BP to a value above the 95th percentile based on age, height, and sex1; PCS low systolic BP, decrease ≥15 mm Hg from baseline low to a value ≤80 mm Hg for children, and decrease ≥20 mm Hg from baseline low to a value ≤90 mm Hg for adolescents; PCS low diastolic BP, decrease ≥10 mm Hg from baseline low to a value ≤50 mm Hg; PCS low pulse, decrease ≥25 from baseline low to a value of <60 for children or decrease ≥15 from baseline low to a value <50 for adolescents; PCS high pulse, increase ≥15 from baseline high to a value >140 for children or increase ≥15 from baseline high to a value >120 for adolescents; PCS weight loss, decrease ≥3.5% from baseline low; Heart rate abnormal high, >130 bpm for children or >110 bpm for adolescents; Heart rate abnormal low, <60 bpm for children or <50 for adolescents; PR interval high, >220 msec; QRS interval high, >120 msec; QTcF high, ≥470 msec for females or ≥450 msec for males; QTcF abnormal increase, >40 msec increase from baseline; QTcF PCS high, >500 msec.
ECG: electrocardiogram, QTcF: QTc Fridericia correction.
National High Blood Pressure Education Program Working Group on High Blood Pressure in Children and Adolescents. 2004
Mean change in systolic BP, diastolic BP, and pulse over the 36 week (acute plus extension phases) longitudinal course of the studies for patients initially randomized to duloxetine or fluoxetine are shown in Figure 1 (panels A, B, and C respectively). For patients initially randomized to duloxetine or fluoxetine, mean increases (<2 mm Hg) in both systolic and diastolic BP were observed at 36 weeks. However, for patients initially randomized to duloxetine, a mean increase in pulse (∼3 bpm) was observed, whereas a mean decrease in pulse (∼1 bpm) was observed at 36 weeks for patients initially randomized to fluoxetine (Fig. 1C).
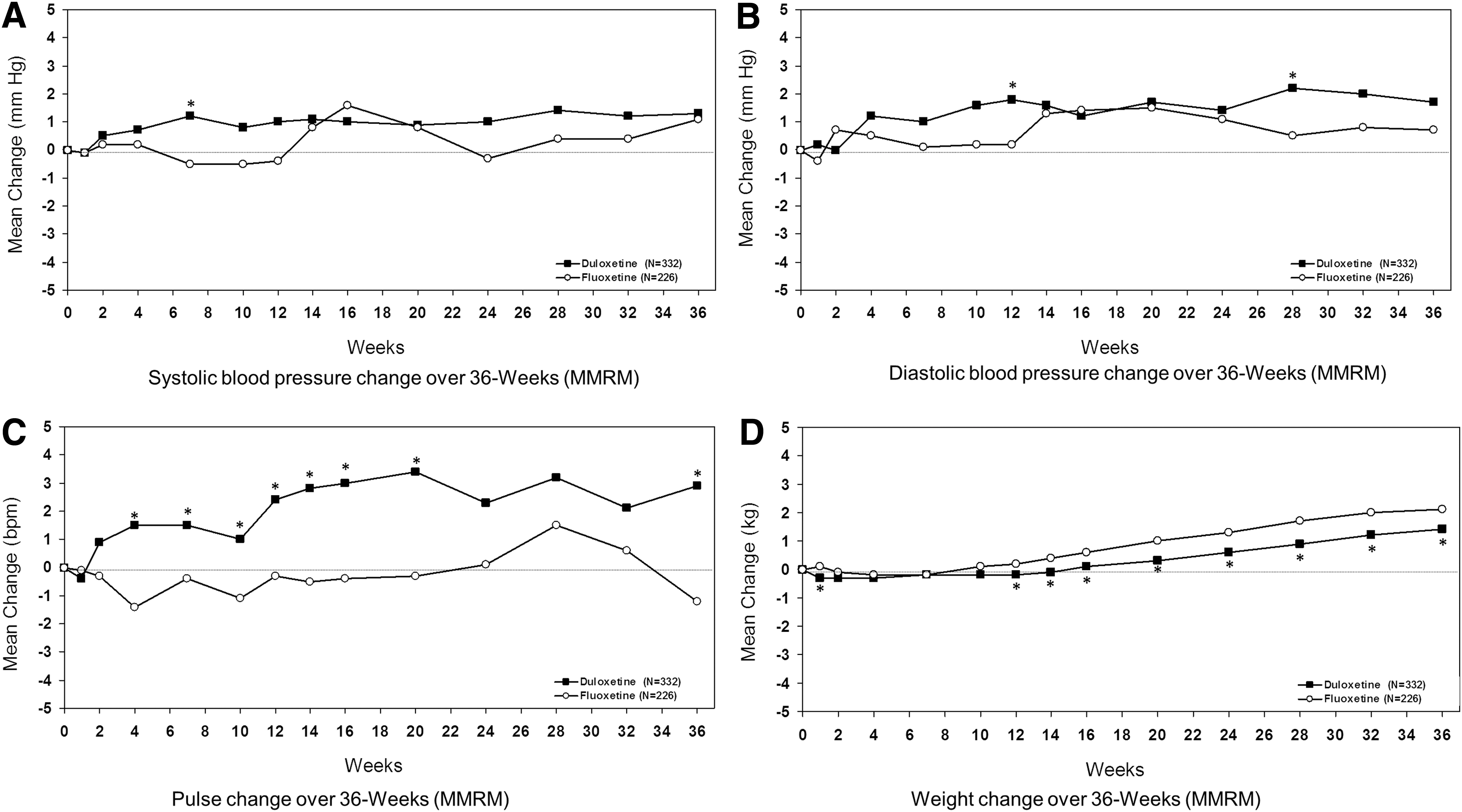
Mean change in systolic blood pressure
Over the 36 week studies, sustained elevations in systolic BP occurred in 4/283 (1.4%) patients initially randomized to duloxetine and 3/199 (1.5%) patients initially randomized to fluoxetine (p=0.9). Sustained elevations in diastolic BP occurred in 5/295 (1.7%) patients initially randomized to duloxetine and 2/205 (1.0%) patients initially randomized to fluoxetine (p=0.4). With the exception of abnormally low heart rate, there were no statistically significant differences between patients initially randomized to duloxetine and those initially randomized to fluoxetine in the incidence of abnormal or PCS electrocardiogram values over the 36 week studies (Table 5).
Laboratory evaluations (hepatic)
Two patients experienced elevated alanine aminotransferase (ALT) level (three or more times the upper limit of normal) during the studies. One duloxetine-treated patient (with abnormal ALT at baseline) had an elevated ALT three or more times the upper limit of normal at the last study visit (week 36), which returned to near normal levels as the patient tapered off of duloxetine per protocol at the completion of the study. One fluoxetine-treated patient had an ALT level of three or more times the upper limit of normal, which returned to normal levels while the patient continued treatment with fluoxetine. No patients met criteria for Hy's Rule (ALT ≥3 times the upper limit of normal and total bilirubin ≥2 times the upper limit of normal [Reuben 2004]).
Growth (weight and height)
The mean baseline body weight of patients in these studies was higher than average compared with age- and sex-matched peers (approximately in the 75th percentile). During the acute treatment phase, there were statistically significant differences between groups in mean weight change and PCS decreases in body weight (Table 5), as well as statistically significant mean decreases in weight z score for both the duloxetine and fluoxetine groups compared with a smaller mean decrease for the placebo group (Table 6). At the last observation in the 10 week acute treatment phase, patients in the duloxetine and placebo groups were at the 75th weight percentile, and patients in the fluoxetine group were at the 70th weight percentile on average (Table 6).
Acute end-point refers to last observation during the 10 week acute treatment phase.
Study end-point refers to last observation over the entire course of the study.
Acute plus extension phase (36 week for weight) and acute plus extension plus taper phases (38 weeks for height).
p≤.05 versus placebo.
p≤.05 duloxetine versus fluoxetine
Mean changes in weight over the 36 week studies are shown in Figure 1D for patients initially randomized to duloxetine or fluoxetine. After the 10 week acute treatment phase, patients initially randomized to duloxetine or fluoxetine gained body weight with mean increases from study baseline of 1.4 kg and 2.1 kg for the duloxetine and fluoxetine groups, respectively, at 36 weeks (p≤0.05 between groups) (Fig. 1D). At last observation in the 36 week studies, PCS decreases in body weight were experienced in 28/332 (8.4%) patients initially randomized to duloxetine and 18/226 (8.0%) patients initially randomized to fluoxetine, with no statistically significant difference between treatment groups. At the last observation during the 36 week study, a statistically significant mean decrease from study baseline in weight z score was observed for patients initially randomized to duloxetine compared with a mean increase in weight z score from study baseline for patients initially randomized to fluoxetine (Table 6). At the last observation in the 36 week studies, patients initially randomized to duloxetine or fluoxetine were on average at the 77th and 74th weight percentiles, respectively (Table 6).
Mean increases in height at the last observation during the 10 week acute treatment phase for duloxetine-, fluoxetine-, and placebo-treated patients were 0.70 cm, 0.56 cm, and 0.65 cm, respectively, with no statistically significant differences between groups. During the acute treatment phase, the same mean decrease in height z score was observed for all treatment groups, with no statistically significant differences among treatment groups (Table 6).
Over the 38 week studies (acute plus extension plus taper phases), patients increased in height by ∼1 cm on average. Over the 38 week studies, a small mean decrease from baseline in height z score was observed for patients initially randomized to duloxetine, compared with a statistically significantly greater mean decrease for those initially randomized to fluoxetine (Table 6). At the last observation during the 38 week studies, patients initially randomized to duloxetine were on average at the 58th height percentile (the same percentile as at study baseline) and patients initially randomized to fluoxetine were on average at the 45th height percentile (a decrease from baseline, Table 6).
Discussion
The safety and efficacy of duloxetine have been studied in ∼8100 adult patients in placebo-controlled studies across five indications overall, and, specifically, in ∼4800 adult patients in placebo-controlled studies of MDD and generalized anxiety disorder (Cymbalta prescribing information 2014). However, duloxetine has been studied in a much smaller number of pediatric patients with MDD (∼500 pediatric patients) from two randomized controlled trials. Data from these two pediatric MDD studies of duloxetine were pooled to allow for an assessment of safety outcomes using the largest possible sample size from similarly designed studies with both a placebo and a fluoxetine control group. The results of the current pooled analyses from duloxetine pediatric MDD studies did not reveal any new safety findings, and the safety and tolerability of duloxetine in the pediatric MDD population was consistent with the known safety and tolerability profile of duloxetine in the adult population (Hudson et al. 2005; Brunton et al. 2010).
During the 10 week acute treatment period, the most frequently reported TEAEs in duloxetine-treated pediatric MDD patients (nausea and other gastrointestinal events, headache, and dizziness) were consistent with those reported in the fluoxetine-treated control group as well as with those observed in the duloxetine-treated adult population (Brunton et al. 2010). It is of note that the overall proportion of patients who reported at least one TEAE did not statistically significantly differ between either of the active drug groups (duloxetine [63%] or fluoxetine [62%]) and the placebo group (62%), nor were there any statistically significant differences between the active drug groups and the placebo group in the proportion of patients reporting TEAEs within the subgroups of children (7–11 years) or adolescents (12–17 years). During the 6 month extension treatment phase, the profile of most frequently reported TEAEs was similar to that observed during the acute treatment phase (nausea and other gastrointestinal events, headache, and dizziness), with an increase in reports of infections (nasopharyngitis and upper respiratory tract infection), which may be anticipated given the longer treatment duration. In addition, the proportion of patients reporting at least one TEAE, as well as the incidence of the most frequently reported individual TEAEs during the extension treatment phase, was greater among those patients transitioned from placebo to duloxetine treatment during the extension phase than among those patients continuing on duloxetine or fluoxetine treatment during the extension phase. This observation is again consistent with the longitudinal profile of TEAEs in the duloxetine-treated adult population, in whom the onset of the most frequent TEAEs occurs early in treatment (Brunton et al. 2010). One SAE of Stevens–Johnson syndrome was reported during the extension phase of one of these studies. Although the case was judged by the investigator to be related to duloxetine exposure (Strawn et al. 2011), this case is atypical, because the patient had only mucosal lesions, and the time to onset was long (138 days).
Suicidal ideation and behavior and nonsuicidal self-injurious behavior information was solicited using the C-SSRS at every visit, and with the exception of one case described in Atkinson et al. (2014), any suicidal ideation or behavior or self-injurious events that were reported spontaneously during AE collection were confirmed to be reported on the C-SSRS.
During the acute treatment phase, there were no statistically significant differences among groups in the incidence of treatment-emergent suicidal ideation, suicidal behavior, or nonsuicidal self-injurious behavior. Among patients who reported suicidal ideation at baseline, >75% in all treatment groups had improvement in suicidal ideation during the acute treatment phase. The frequency of suicide-related events (via C-SSRS) for duloxetine- and fluoxetine-treated patients as well as placebo-treated patients during the acute treatment phase was consistent with that reported in the antidepressant group (0–8% for suicidal ideation and behavior) in Hammad et al. (2006).
Placing the longer-term results into context is more difficult than for acute results, because there have been relatively few longer-term studies of antidepressants in pediatric patients with MDD. The Treatment for Adolescents with Depression Study (TADS), however, was one such longer-term study that used methodology for assessing suicidal events that was similar (though not identical) to the C-SSRS methodology used in the current analysis. Overall in the TADS study, suicidal behavior (suicide attempt) was reported in 7/109 (6.4%) antidepressant monotherapy patients over the 36 weeks of treatment, with two incidents being reported during the 12 week acute treatment and five additional incidents being reported during the 24 week extension treatment (Emslie et al. 2006; Vitiello et al. 2009). In the current pooled analysis, for those patients initially randomized to duloxetine, the frequency of suicidal behavior during the 36 weeks of treatment was 6/333 (1.8%) and for patients initially randomized to fluoxetine it was 3/225 (1.3%), which was less than that reported in TADS (6.4%). In addition, these studies were among the few studies to measure improvement in suicidal ideation over longer time frames, with >75% of patients initially randomized to duloxetine or fluoxetine who had suicidal ideation at baseline reporting a reduction in the severity of suicidal ideation at end-point during the 36 week studies.
With regard to cardiovascular outcomes, the incidence of sustained elevation in BP for three consecutive visits during the 36 week studies did not statistically differ between patients initially randomized to duloxetine and those initially randomized to fluoxetine. Additionally, laboratory and electrocardiogram results did not reveal any new safety findings for either duloxetine or fluoxetine. There were no statistically significant differences for either active drug group (duloxetine or fluoxetine) compared with the placebo group in mean change in pulse or BP during the acute treatment phase; and over the 36-week longitudinal course of the studies, mean increases in BP and pulse for duloxetine-treated pediatric patients were consistent with those observed for duloxetine-treated adult patients (Hudson et al. 2005). This greater increase from baseline in BP and pulse for duloxetine-treated pediatric patients compared with minimal change from baseline for fluoxetine-treated pediatric patients is consistent with the pharmacological action of duloxetine (primary action on both serotonergic and noradrenergic neurotransmission) and fluoxetine (primary action on serotonergic neurotransmission). Although no statistically significant differences in BP and pulse were observed between the duloxetine and placebo groups during the 10 week acute treatment phase, mean increases were observed for duloxetine-treated patients relative to placebo-treated patients; therefore, BP should be measured prior to initiating treatment and periodically measured throughout treatment, and it is reasonable to monitor pulse during treatment for medications that may increase noradrenergic neurotransmission.
Potential medication effects on growth are a topic of interest in pediatric patients. Specifically, in the current analysis, given the statistically greater incidence of PCS weight loss in both duloxetine- and fluoxetine-treated patients compared with the placebo group, there is a question if the observed weight loss is associated with a decrease in the growth rate in height. First, with regard to weight, patients in the current analysis were at the 75th percentile on average at baseline compared with age- and sex-matched peers; therefore, even though an average decrease in weight percentile of 2–3% was observed during the acute treatment phase, patients were at the 70th percentile or greater on average after acute treatment. Additionally, during the extension phase, patients trended toward weight recovery to their baseline weight percentile.
With regard to height, there were no statistically significant differences between treatment groups during the acute treatment phase, with similar mean decreases in height percentile (∼1% decrease) for both active drug groups and the placebo group. Over the 38 week studies, mean decreases in height percentile were observed for the duloxetine group (0.4% decrease) and the fluoxetine group (2% decrease), and at end-point, patients in the duloxetine group were at the 58th percentile on average, and patients in the fluoxetine group were at the 45th percentile on average. Although a greater initial mean decrease in weight was observed for duloxetine-treated patients than for fluoxetine-treated patients, this did not translate into a continued decrease in height percentile over the extension treatment phase for the duloxetine group, but rather a gradual recovery toward baseline height percentile over the 38 week course of the studies was observed. Nevertheless, as PCS weight loss was observed for some duloxetine- and fluoxetine-treated patients, regular monitoring of growth (weight and height) should be performed in pediatric patients treated with duloxetine or fluoxetine.
Limitations
Several limitations of the current analysis must be taken into account when interpreting the results. First, this analysis pooled results from fixed and flexible dose drug treatment arms; therefore, a clear assessment of dose effects on safety parameters could not be made. However, the best approach to assess dose–response relationships is through fixed dose studies, and one of the studies in the current pooled analysis was a fixed dose study that included more information on dose effects (Emslie et al. 2014). Second, for safety parameters such as growth, a 38 week study is not likely to be sufficient to fully characterize longitudinal safety outcomes. Third, in the two pediatric MDD studies included in this analysis, neither the investigational drug (duloxetine) nor the active control (fluoxetine) demonstrated a statistically significant separation from placebo on the primary efficacy analysis, thus rendering the studies inconclusive (Atkinson et al. 2014; Emslie et al. 2014); therefore, the safety results presented herein cannot be associated with efficacious treatment of MDD in the pediatric patient population. However, the drug doses used for these studies were in the approved range for pediatric use for fluoxetine and in the approved dose range in adults for duloxetine. Finally, although these studies included double-blind, fluoxetine-controlled extension phases, the placebo-controlled phases addressed only acute efficacy (10 weeks of treatment); therefore, longer-term safety outcomes for patients receiving antidepressant treatment could not be evaluated against a no-antidepressant control group in the current analysis.
Conclusions
The current pooled analysis of two clinical trials provides a larger database in which to assess safety outcomes than in smaller individual studies. Results from this analysis were consistent with the known safety and tolerability profile of duloxetine. The nature and profile of TEAEs reported for duloxetine-treated children and adolescents were generally similar to those reported in adult patients with MDD (Brunton et al. 2010). As assessed by the C-SSRS, treatment-emergent suicidal ideation occurred in ∼6–8% of duloxetine- or fluoxetine-treated patients and ∼8% of placebo-treated patients during acute treatment, with no statistically significant differences between either of the active drug groups and the placebo group, and this rate is consistent with the findings for antidepressant treatment arms reported in Hammad et al. 2006. Finally, no new safety findings were identified based on vital signs, electrocardiogram, or laboratory analyses.
Clinical significance
Results from this pooled analysis of two studies were consistent with the known safety and tolerability profile of duloxetine.
Footnotes
Acknowledgments
The authors thank the investigators and their staff, the patients and their families, and the clinical operations staff and statistical analysts of the Duloxetine Antidepressant Team for their excellent implementation of the trials represented in this analysis. The authors also thank Joseph Giaconia and Elizabeth Gardner of INC Research (Raleigh, NC) for their writing and editorial support.
Disclosures
Graham Emslie has received research support from Biobehavioral Diagnostic Inc., BioMarin, Duke University, Eli Lilly, Forest Laboratories, GlaxoSmithKline, Mylan, the National Institute of Mental Health (NIMH), and Somerset; has served as a consultant for Alkermes, Inc., Allergan, NCS Pearson (previously BioBehavioral Diagnostics Inc.), Bristol-Myers Squibb, Eli Lilly, Forest Laboratories, GlaxoSmithKline, INC Research Inc., Lundbeck, Merck, Pfizer, Seaside Therapeutics, Shire, the Texas Department of State Health Services, University of Miami, Valeant, and Wyeth; and was on the Speakers Bureau for Forest Laboratories. Now an emeritus faculty member, John S. March has no established relationships with the pharmaceutical industry. Dr. March has served as a consultant or scientific advisor to Attention Therapeutics, Bristol Myers Squibb, Eli Lilly and Company, and Pfizer; received study drugs for a NIMH‐funded study from Eli Lilly and Company and Pfizer; is an equity holder in MedAvante; and receives royalties from Guilford Press, Multihealth Systems, and Oxford University Press. Dr. March has received research support from National Institute on Drug Abuse (NIDA), NIMH, and Pfizer. Dr. March has not engaged in promotional work (e.g., being part of a speakers bureau or training) for>15 years. Dr. Wells has served as a consultant for Eli Lilly and Company and Merck and serves on a data and safety monitoring board (DSMB) for Novartis. Apurva Prakash, Qi Zhang, Beth Pangallo, and Mark Bangs are all employees of and own stock or equity in Eli Lilly and Company.