
Abstract
Objective:
To assess activating and tranquilizing effects of second-generation antipsychotics (SGAs) in youth.
Methods:
As part of the naturalistic inception cohort study, “Second-generation Antipsychotic Treatment Indication, Effectiveness and Tolerability in Youth (SATIETY),” subjective ratings of activating and tranquilizing symptoms were obtained monthly for 3 months from antipsychotic-naïve youth initiating SGAs using the Treatment Emergent Symptoms Scale (TESS). Discontinuation rates, and TESS-reported symptom rates, and severity were related to clinical and treatment parameters. Two compound measures of TESS were defined: presence of any daytime activating (ACTIVATION+) and sedating symptoms (SEDATION+).
Results:
In 327 antipsychotic-naïve youth originally initiating the four studied SGAs, discontinuation due to sedation was marginally highest with quetiapine (13.0%) followed by olanzapine (7.3%), risperidone (4.2%), and aripiprazole (2.0%) (p = 0.056). Two hundred fifty-seven antipsychotic-naïve youth (13.8 ± 3.6 years, male = 57.8%) initiated aripiprazole (n = 40), olanzapine (n = 45), quetiapine (n = 36), or risperidone (n = 135) and completed ≥1 postbaseline follow-up visit. Baseline prevalence of ACTIVATION+ (39.9%) or SEDATION+ (54.1%) did not differ between SGAs. Rates of both compound measures changed significantly over time (decrease for ACTIVATION+, p = 0.0002; increase for SEDATION+, p < 0.0001) with slight differences between SGAs, explained by lower rates of ACTIVATION+ with olanzapine (p = 0.002) and slightly higher rates of ACTIVATION+ with aripiprazole (p = 0.018) during follow-up, and lower rates of SEDATION+ with aripiprazole (p = 0.018). All four SGAs reduced insomnia (p = 0.001) and increased hypersomnia (p < 0.001). Postbaseline prevalence of drowsiness, the most frequent, but mild TESS complaint was 85%, without SGA differences. Younger age was associated with activating symptoms, higher age with sedating symptoms, and lower baseline functioning increased both. Psychomotor retardation rates were high in subjects with schizophrenia-spectrum disorders, whereas stimulant comedication was associated with psychomotor activation, regardless of diagnosis.
Conclusions:
Although small SGA-specific differences in activating/sedating compound side effect measures were noted, independent predictors of single TESS ratings included clinical parameters, rather than specific SGAs, suggesting a need for carefully individualized treatment strategies.
Introduction
S
Registration trials reported increased, but variable rates of somnolence or sedation relative to placebo for all SGAs in pediatric patients with schizophrenia and bipolar disorder [rates for aripiprazole 11%–26% (Findling et al. 2008, 2009); for olanzapine 5%–23% (Tohen et al. 2007; Kryzhanovskaya et al. 2009); for quetiapine 28%–29% (Findling et al. 2012; Pathak et al. 2013); and for risperidone 12%–22% (Haas et al. 2009a, 2009b)].
Tolerance for these adverse effects often develops during extended treatment periods, but persisting high rates have nevertheless been reported (Findling et al. 2013; Flank et al. 2014). Rates of restlessness, agitation, or psychomotor activation as adverse effect have reportedly been slightly to much lower [rates for aripiprazole 8%–12% (Findling et al. 2008, 2009); for olanzapine <5% (Kryzhanovskaya et al. 2009); for quetiapine <5%–8% (Findling et al. 2012; Pathak et al. 2013); and for risperidone 11%–15% (Haas et al. 2009a, 2009b)]; at times being even lower than in the placebo-treated comparison group (Findling et al. 2012).
In contrast to sedating effects, activating effects cannot be readily distinguished from an insufficiently improved or exacerbated psychiatric disorder; that is, a lack of efficacy, rather than a side effect. In registration trials, rescue medication is often allowed for activating symptoms. Nevertheless, both sedating and activating effects can be among the reasons leading to study discontinuation [for sedation (Findling et al. 2009; Haas et al. 2009a, 2009b); and for agitation (Findling et al. 2012)].
Despite their relevance for academic and social achievement in youth, tranquilizing and activating adverse effects of SGAs have not been compared directly in pediatric patients. Meta-analyses in adults suggested less sedation with risperidone relative to quetiapine, but not relative to other SGAs (Komossa et al. 2011). Olanzapine was deemed to convey comparable rates of sedation relative to quetiapine or risperidone (Komossa et al. 2010), but a reduced risk of sedation was found for aripiprazole relative to olanzapine (Komossa et al. 2010).
Treatment decisions during standard clinical care are being informed by these data, but populations in randomized controlled trials may not be generalizable and data from patients with bipolar disorder and schizophrenia may not apply to youth with other disorders, which is relevant given increased off-label use of antipsychotics. Moreover, double-blind, randomized controlled trials often use unsolicited reporting, instead of proactive, scale-based questioning about side effects and may thus underestimate the subjective side-effect burden.
Conversely, clinicians in routine care can readily react to complaints about side effects, in particular if these are deemed dose-dependent, while this is not an option in fixed-dose studies, in which adverse events could thus occur more frequently or be more severe or prolonged. Yet, the reporting of one rate for presence or absence of a certain adverse effect during the entire study, as typically done in randomized trials, obscures the clinically very relevant information of whether or not tolerance developed or whether dose adaptations could attenuate the side effect. Prospectively assessed rates of sedating and activating side effects in youth during routine psychiatric care with different SGAs are currently lacking. However, such data can add to the knowledge gained from controlled trials, by adding information on the frequency, severity, and manageability of these side effects under usual care conditions, helping to further evaluate the safety profiles of these broadly used medications.
In this study, we used the subjectively reported treatment emergent symptoms scale (TESS) (Guy 1976) to assess the dimensions of sedation and activation during SGA treatment in routine child and adolescent psychiatry care of antipsychotic-naïve youth to (1) prospectively quantify prevalence rates of drug-induced changes in vigilance during the first 3 months of naturalistic use of SGAs and (2) identify risk profiles for these subjective adverse effects in this population.
Materials and Methods
Study setting and design
Data were collected as part of the Second-Generation Antipsychotic Treatment Indication, Effectiveness and Tolerability in Youth (SATIETY) study (Correll et al. 2009), a prospective, naturalistic inception cohort study of antipsychotic-naïve youth started on SGAs to treat psychotic, mood, or aggression-spectrum disorders.
Subjects were recruited through the Zucker Hillside Hospital (Queens, NY), and informed consent or assent (in minors) was obtained from all participants or guardians under a protocol approved by the North Shore-Long Island Jewish Health System Institutional Review Board. All procedures were in accordance with the ethical standards on human experimentation (institutional and national) and with the Helsinki Declaration of 1975/2000. For this analysis, data were analyzed from patients recruited from inpatient and outpatient services from December 2001 to September 2007 (Correll et al. 2009).
Subjects
Inclusion criteria were as follows: age 4–19 years; ≤1 week of lifetime antipsychotic exposure; psychiatric illness prompting antipsychotic initiation; and consent, baseline anthropometric, biochemical assessments obtained within ≤7 days of antipsychotic initiation. Exclusion criteria were as follows: treatment with >1 antipsychotic; active/past eating disorder; biochemical evidence of thyroid dysfunction; acute medical disorders; pregnancy/breastfeeding; and leaving the catchment area within <4 weeks. Diagnoses and treatment decisions were made as part of clinical care by board-certified child and adolescent psychiatrists according to Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV) criteria and recorded in the clinical chart. Psychiatric diagnoses and past treatment history were further assessed by the study investigators through chart review, discussion with treating clinicians, and clinical interview of the patient and/or caregiver.
Treatment
Patients received clinician's choice antipsychotic treatment. Informed consent/assent was obtained only after the antipsychotic choice was made. Dosing, comedications, and treatment changes were based solely on clinical need, but were documented during each study visit. As a measure of exposure, the maximum daily dose per time frame was documented. As part of the broader assessments within the context of the SATIETY study (for details see Correll et al. 2009) blood samples, including blood levels of antipsychotics, were obtained at each study visit. To facilitate the assessment of dose–side effect analyses across SGA subgroups, we converted SGA doses into chlorpromazine equivalent (CPZE) doses (Andreasen et al. 2010).
Outcomes and assessments
Subjective reports of adverse events were recorded at baseline and during monthly visits (week 4, 8, 12) by the investigators with a modified version of the “Treatment Emergent Symptoms Scale” (TESS) (Guy 1976). Answers on this scale are obtained in a semistructured interview of the patient (whenever possible in the presence of a caregiver who could provide additional input) with systematic questioning about the presence or absence of a given side effect and about the severity hereof. Symptoms are coded on a four-point Likert scale as absent, mild, moderate, or severe.
Overall, the scale consists of 34 items, covering all organ systems systematically. For this study, daytime tranquilizing effects were assessed with the items psychomotor retardation, malaise, and drowsiness, while daytime activating effects were assessed with the items agitation, restlessness, and psychomotor activation. In addition, sleep-related symptoms of hypersomnia and insomnia were recorded, considering cumulative sleep duration during a 24-hour period compared to the usual sleep duration. Furthermore, for the purpose of these analyses, we defined two compound variables from the daytime TESS ratings as the coprimary outcome variables: • The parameter summarizing the daytime activating symptoms was coined ACTIVATION+. ACTIVATION+ was coded as 0 if no agitation, restlessness, or psychomotor activation was reported on the TESS and as 1 if any item of agitation, restlessness, or psychomotor activation had been rated with ≥1 (≥mild). • The parameter summarizing the daytime sedating symptoms was coined SEDATION+. SEDATION+ was coded as 0 if no malaise, drowsiness, or psychomotor retardation was reported on the TESS and as 1 if any item of malaise, drowsiness, or psychomotor retardation had been rated with ≥1 (≥mild).
Data analysis
Visits from the time of absent antipsychotic levels or reported nonadherence (by patient or caregiver) were censored. We used logistic regression to estimate the effect of time and antipsychotic subgroup on the coprimary outcome parameters (ACTIVATION+; SEDATION+).
We also analyzed single item TESS ratings as secondary outcome parameters. Comparison of rates between baseline and each follow-up time point, and group comparisons were performed using chi-square (χ 2) tests.
Group comparisons of symptom rates and severity were performed within time point and for postbaseline period prevalences. Severity changes of TESS ratings were determined using the Wilcoxon signed rank test. We did not report incidence rates (i.e., only new symptoms occurring after baseline) as not all patients were assessed before the first dose was received.
Interactions of medication dose and TESS symptom severity were estimated using ordinal logistic regression. Dose comparisons in groups with versus without specific TESS symptoms were calculated using Wilcoxon rank sum tests.
To test for the association of clinical factors and the period prevalence of TESS symptoms ratings (absent vs. present), we used multivariable nominal logistic regression with backward elimination of nonsignificant factors (p > 0.05) starting with the factors antipsychotic subgroup, CPZE maximum dose during the 3-month period, primary psychiatric diagnosis, age, sex, inpatient/outpatient status, and baseline functioning (CGAS baseline score; all as listed in Table 1). The models also included a variable coding for concomitant stimulant use for all analyses on activating side effects or a variable coding for concomitant mood stabilizer use for all analyses on sedating side effects.
Bolded p-values: <0.05.
ADHD, attention-deficit/hyperactivity disorder; CD, conduct disorder; CGAS, Children's Global Assessment Scale; CPZE, chlorpromazine equivalent; ICD, impulse control disorder; IED, impulsive explosive disorder; ODD, oppositional defiant disorder; SD, standard deviation.
To exclude a bias of not entering patients into the analyses, who discontinued treatment because of the adverse effects under investigation, we also assessed and compared dropout rates due to any of the investigated adverse effect outcomes in the full intent-to-treat (ITT) sample. Data were analyzed using JMP 12.0 (SAS Institute, Cary, NC); tests were two-sided and alpha was set at 0.05.
Results
Study population
Of 505 antipsychotic-naïve pediatric patients, 338 (66.9%) were consented into the SATIETY study. After excluding 5 patients who did not start the antipsychotic treatment and 6 patients who started on ziprasidone due to the small sample size, 327 antipsychotic-naïve patients (mean age = 14.0 ± 3.5 years, 56.2% male, 47.1% White, 71.0% inpatients; mood-spectrum disorders: 48.1%, schizophrenia-spectrum disorders: 31.0%, disruptive behavior-spectrum disorders: 20.9%) comprised the ITT sample. Furthermore, 10 patients were nonadherent before the first 4-week visit (assessed by patient/caregiver report and antipsychotic blood level), and 60 (17.9%) did not undergo a postbaseline assessment due to early drop out, yielding 257 (76.0%) patients with confirmed antipsychotic adherence and at least one postbaseline assessment within 3 months in the final baseline modified ITT (mITT) sample.
In the mITT sample of 257 antipsychotic-naïve youth (mean age = 13.8 ± 3.6 years, 57.8% male, 49.2% White, 68.1% inpatients) primary diagnoses included mood-spectrum disorders (45.9%), disruptive behavior-spectrum disorders (22.6%), and schizophrenia-spectrum disorders (31.5%). Subjects were started on aripiprazole (n = 41), olanzapine (n = 45), quetiapine (n = 36), or risperidone (n = 135) as per clinical treatment choice independent of the study team. As expected in a naturalistic study, antipsychotic treatment groups differed with regard to several aspects (Table 1). Subjects starting as inpatients were overrepresented in the olanzapine group, as were males. This group also showed the lowest baseline functioning and, together with the quetiapine treatment group, the highest frequency of mood stabilizer comedication.
Reasons for drop out (ITT sample)
Of all examined activating or tranquilizing adverse effects, only sedation was one of the reasons for early treatment discontinuation. In the entire ITT sample, there was a nonsignificant trend (p = 0.059) toward more discontinuations due to sedation in patients treated with quetiapine (7/54 = 13.0%) compared to the other SGAs, that is, olanzapine (4/55 = 7.3%), risperidone (7/167 = 4.2%), and aripiprazole (1/51 = 2.0%). Similarly, groups did not differ when comparing the frequency of sedation in the 138 patients, who discontinued SGAs prematurely for any reason, that is, quetiapine (7/27 = 25.9%), olanzapine (4/19 = 21.1%), risperidone (7/75 = 9.3%), and aripiprazole (1/17 = 5.9%) (p = 0.096).
Primary outcome: activating and tranquilizing effects (mITT sample)
The presence of any daytime activating or any daytime sedating symptom as summarized in the parameters ACTIVATION+ (present in 39.9%) and SEDATION+ (present in 54.1%) did not differ between antipsychotic subgroups at baseline (p = 0.44 and p = 0.16, respectively).
The logistic fit (full model, χ 2 = 36.42; p < 0.0001) for ACTIVATION+ showed a significant effect of time (χ 2 = 20.07; p = 0.0002) and a significant effect of antipsychotic group (χ 2 = 16.28; p = 0.001); with a significant difference in ACTIVATION+ from baseline to week 4 (p = 0.02), from week 4 to 8 (p = 0.04), and a plateau thereafter (p = 0.14). The effect of antipsychotic group was explained by relatively lower rates of ACTIVATION+ during follow-up with olanzapine (χ 2 = 13.98; p = 0.002) and slightly higher rates of ACTIVATION+ with aripiprazole (χ 2 = 5.61; p = 0.018).
The logistic fit (full model, χ 2 = 39.40; p < 0.0001) for SEDATION+ showed a highly significant effect of time (χ 2 = 31.15; p < 0.0001) and a slight effect of antipsychotic group (χ 2 = 8.29; p = 0.040); with significant differences in SEDATION+ from baseline to week 4 (p > 0.0001), from week 4 to 8 (p = 0.001), and a plateau thereafter (p = 0.12). The effect of antipsychotic group was explained by relatively lower rates of SEDATION+ during follow-up with aripiprazole (χ 2 = 5.65; p = 0.018).
Secondary outcomes: prevalence and severity of single TESS items over time
Prevalence rates of single TESS items and postbaseline period prevalences of single TESS items, as well as group comparisons hereof are provided in Table 2. Detailed information on the severity of subjective ratings, reflected as mean scores of TESS ratings at week 4, 8, and 12, as well as tests for changes of these measures and group comparisons of the severity ratings for single items are provided in Table 3.
Bold p-values: p < 0.05
SGAs, second-generation antipsychotics.
Bolded p-values: <0.05.
SD, standard deviation.
Rates of agitation were low (13.6% postbaseline period prevalence, Table 2) and mild (Table 3), both at baseline as well as during follow-up. For the mITT sample, there was a slight decrease in the rate of subjects experiencing agitation at week 8; otherwise rates remained constant.
Restlessness decreased significantly during follow-up in the entire mITT sample. There were no significant group differences in the rates (Table 2) and severity ratings (Table 3), and the within-group comparisons over time showed a significant reduction of restlessness rates and severity ratings with risperidone, olanzapine, and less so with quetiapine (Fig. 1). These changes were absent in aripiprazole.
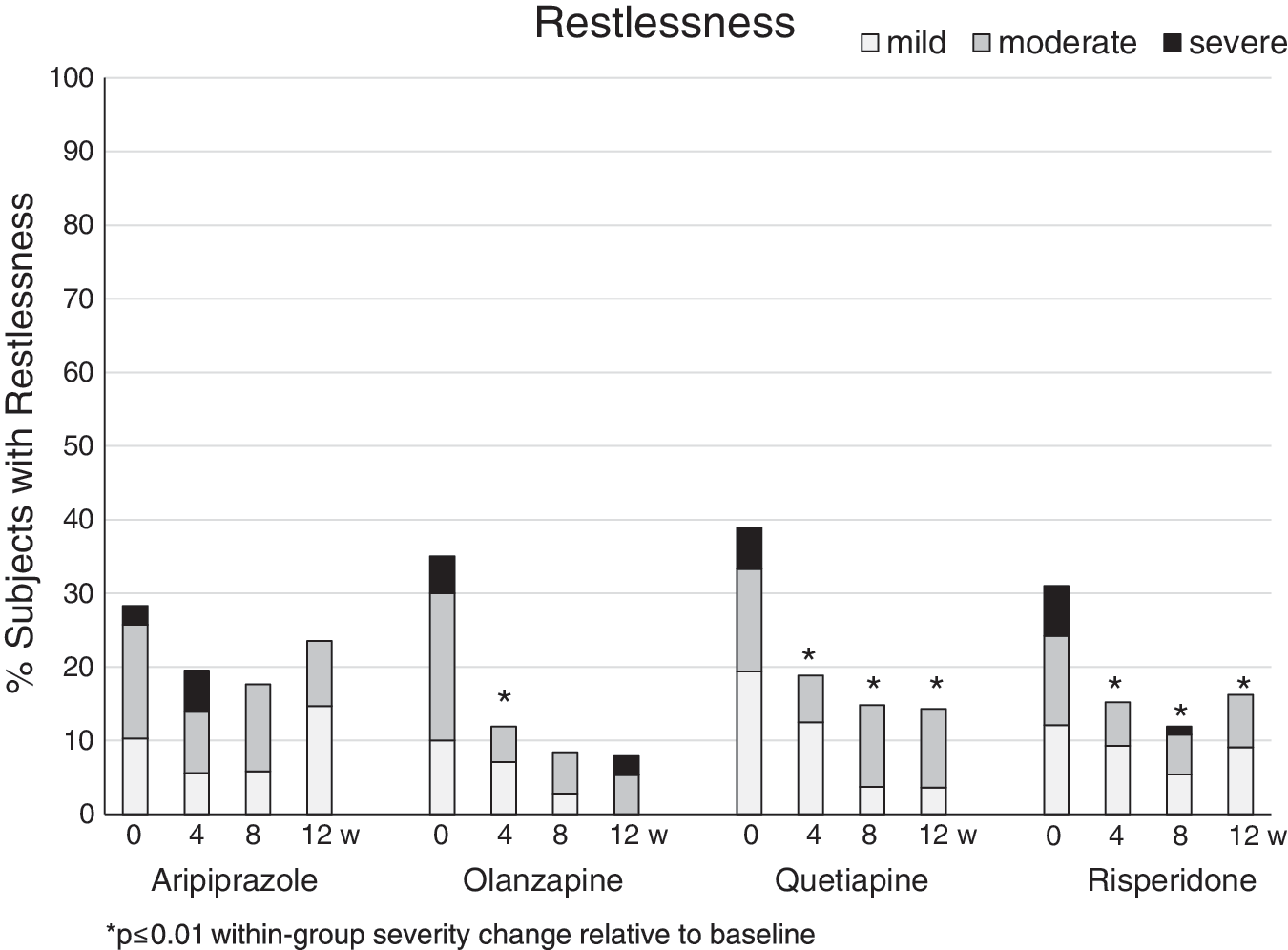
Severity rates of restlessness within the four second-generation antipsychotic treatment groups aripiprazole, quetiapine, olanzapine, and risperidone (left to right) at baseline and week 4, 8, and 12 (left to right). Grey scales indicate symptom severity as subjectively rated on the Treatment Emergent Symptom Scale from mild (light grey), to moderate (dark grey) to severe (black).
The rate of subjects experiencing psychomotor activation as a side effect did not change over time, and no group difference in these rates was present at any time point (Table 2). The severity of psychomotor activation ratings, however, decreased slightly from baseline to week 4 in the mITT sample, but mainly so in the aripiprazole group (Table 3).
Even though most sedating symptoms were frequent in the mITT sample, they were predominantly of mild severity. In particular, drowsiness was the most frequently reported daytime complaint, both at baseline (44.3%) as well as cumulatively during the 12-week observation period (84.9%; Table 2). Moreover, there was a significant change of sleep patterns in the full mITT sample with an increase of hypersomnia and a decrease of insomnia, both with regard to the rate of complaints as well as with regard to reported severity. Except for slightly increasing severity ratings of hypersomnia at week 4 in olanzapine, this change in sleep pattern was not different between antipsychotic groups. Specific changes of symptom rates and severity ratings are detailed below.
In summary, significant changes were most pronounced in comparisons of baseline versus week 4, but values continued to differ from baseline, without returning to baseline values. Thus, on the group level, tolerance development was not observed.
Malaise remained constant over time, without group differences in these rates or severity ratings, except for a small dip with risperidone at week 12.
Importantly, despite being already frequent at baseline, rates (Table 2) and severity (Table 3) of drowsiness ratings increased significantly over time. Increases in the severity of drowsiness were particularly pronounced with olanzapine, but absent with quetiapine, without reaching statistical significance in between-group analyses (Fig. 2).

Severity rates of drowsiness within the four second-generation antipsychotic treatment groups at baseline and week 4, 8 and 12 (left to right). Grey scales indicate symptom severity as subjectively rated on the Treatment Emergent Symptom Scale from mild (light grey), to moderate (dark grey) to severe (black).
The proportion of subjects experiencing psychomotor retardation as well as severity ratings of psychomotor retardation remained constant over time in the entire mITT sample. However, the rate of psychomotor retardation significantly dropped from 16.4% to 0% in the quetiapine treatment group (at week 8; p = 0.035). Accordingly, a significant group difference (p = 0.028) in the postbaseline period prevalence of this side effect was noted favoring quetiapine. Similarly, low rates of psychomotor retardation were also seen in the risperidone group. By contrast, severity ratings of psychomotor retardation increased slightly in the olanzapine group at week 4.
Dose–side effect associations
No significant associations were found in logistic fits of ACTIVATION+ or SEDATION+ with the maximum daily CPZE dose at week 4, 8, or 12. Similarly, there was no association of single TESS items with the maximum daily CPZE dose at week 4 or 12 in the mITT sample (p > 0.05 for all tests).
Significant dose–side effect associations were, however, found in the risperidone group, with higher doses being present in subjects reporting sedating symptoms and lower doses in subjects reporting activating symptoms (Table 4).
Bolded p-values: <0.05.
PM, psychomotor.
Logistic fits with the severity levels of symptoms showed that lower doses were significantly associated with higher subjective severity ratings of psychomotor activation at week 4 (χ 2 = 9.4; p = 0.002; n = 118). Higher doses were significantly associated with higher subjective ratings of psychomotor retardation (χ 2 = 11.19; p = 0.0008; n = 118) at week 4. Similar associations were present at week 12 (psychomotor activation; χ 2 = 6.5; p = 0.011; psychomotor retardation; χ 2 = 5.6; p = 0.017; n = 102).
Risk profiles for specific symptoms
Significant models were calculated for the emergence (postbaseline period prevalence) of restlessness, psychomotor activation, drowsiness, and psychomotor retardation (Table 5). By contrast, no significant models could be constructed for agitation, malaise, insomnia, and hypersomnia (p > 0.05 for the full model).
CGAS, Children's Global Assessment scale; CI, confidence interval; NPV, negative predictive value; OR, odds ratio; per unit increase for continuous variables; PPV, positive predictive value; RIS, risperidone.
Generally, younger age was an independent predictor for activating symptoms, whereas relatively older age was a predictor of sedating symptoms, each while controlling for the other factors in the model (for details see Table 5). Moreover, lower baseline functioning contributed to the likelihood of both activating and sedating symptoms. Importantly, antipsychotic treatment groups did not separate as independent predictors, except for aripiprazole, which significantly reduced the risk of psychomotor retardation.
In addition, the following factors each contributed significantly to the likelihood of experiencing specific complaints, while controlling for the other factors in the model:
Outpatient status significantly increased the likelihood to report restlessness.
Stimulant comedication and outpatient status increased the likelihood to report psychomotor activation. Stimulant comedication was only partly separable from the diagnosis of attention-deficit/hyperactivity disorder (ADHD; 53.7% of subjects with ADHD did not receive stimulants), which had, however, less predictive power in the explanatory model (full model as detailed in Table 5, but with ADHD instead of stimulant χ 2 = 70.4; p > 0.0001; ADHD: χ 2 = 10.5; p = 0.002).
Psychomotor retardation was less likely in subjects with a primary diagnosis of disruptive behavior-spectrum disorders relative to subjects with schizophrenia-spectrum disorders, and in females.
Drowsiness was solely explained by the factors clinical functioning and age, as specified above.
Discussion
This is the first prospective naturalistic observational study on tranquilizing and activating adverse effects of SGAs, describing and comparing the effects of the four most prescribed SGAs in antipsychotic-naïve children and adolescents treated by clinician's choice under real-world conditions.
All four SGAs significantly affected sleep patterns, with a decrease of the undesirable insomnia, but an increase of equally undesirable hypersomnia over time. Additional daytime tranquilizing symptoms were frequent (affecting 85% cumulatively), but generally mild. Drowsiness, as the most frequent complaint, rose to severe or moderate severity in a third of subjects during the first month of treatment. In general, all daytime tranquilizing effects, although already frequent at baseline, increased significantly throughout the observation period without wide-spread development of tolerance. This tranquilizing effect in youth was homogenously found in all antipsychotic subgroups, except for a slight attenuation of sedating effects with aripiprazole.
Activating symptoms were generally less frequent and significantly decreased with time. This decrease was pronounced with olanzapine, and absent with aripiprazole. The largest changes in both, activating or sedating symptoms, were observed in the first 4 weeks, with relatively stable measures past 8 weeks, indicating little, if any, development of tolerance. Although quetiapine, followed by olanzapine, caused numerically more discontinuation due to sedation than risperidone and aripiprazole, this difference was not statistically significant, and none of the other tranquilizing or sedating adverse effects led to treatment discontinuation.
These results largely match the side-effect profiles as seen in data in adults (Leucht et al. 2013), with tranquilizing effects being more prominent with SGAs that are highly antihistaminergic, such as olanzapine and quetiapine, and with restlessness to be most prominent with the partial dopamine agonist aripiprazole. In general, youth have been reported in indirect comparisons to be more vulnerable to experiencing/reporting sedation, somnolence or drowsiness than adults (Correll et al. 2006), and our data confirm these earlier observations.
For example, while adults taking aripiprazole reported sedation rates of 11% (Marder et al. 2003; McQuade et al. 2004), in pediatric patients, sedation ranged from 0% to 33% (Barzman et al. 2004; Biederman et al. 2005; Findling et al. 2008, 2009). Olanzapine has been shown to cause sedation in 25%–39% of adults (Conley and Mahmoud 2001; McQuade et al. 2004), compared to high pediatric rates of 44%–94% in some studies (Kumra et al. 1998; Frazier et al. 2001; Findling et al. 2003; Sikich et al. 2004), being lower in others (5%–23%) (Tohen et al. 2007; Kryzhanovskaya et al. 2009). Between 18% and 37% of adults taking risperidone experienced sedation (Azorin et al. 2001; Conley and Mahmoud 2001; Addington et al. 2004), whereas 29%–89% of children reported feeling sedated (Turgay et al. 2002; Aman et al. 2004; Shea et al. 2004; Sikich et al. 2004). With quetiapine, sedation in adults ranged from 34% to 43% (Vieta et al. 2002; Yatham et al. 2004) compared to rates as high as 25%–80% in youth (Delbello et al. 2002; McConville et al. 2003; Mukaddes and Abali 2003; Findling et al. 2012; Pathak et al. 2013).
In contrast, rates in our study were even higher with the four examined SGAs, ranging between 50% and 80% when proactive elicitation, rather than when spontaneous side-effect reporting was employed.
However, spontaneous side-effect reports as documented in registration trials may reflect more moderate to severe symptom severity, while the prevalences in our study predominantly reflect rates of at least mild severity, obtained by systematic questioning. Rates of moderate-to-severe tranquilizing symptom severity observed in our study were comparable to those reported from registration trials, with peaks at week 4 of 33% for drowsiness (Fig. 2), 16% for psychomotor retardation, and 11% for malaise (all in moderate-to-severe severity). Nevertheless, these rates appear concerning as treating clinicians were not blind to treatment status and were thus able to balance the benefit–side effect ratio with dose adaptations. This active side effect management is also likely to have attenuated clearer distinctions of antipsychotic treatment groups with regard to tranquilizing symptoms.
Earlier studies, across a variety of diagnoses showed that the numbers needed to harm compared to placebo for sedation, somnolence, or drowsiness varied from 5–6 for quetiapine and olanzapine, to 3–13 for risperidone, and to 5–20 for aripiprazole (Correll 2008). Similarly, the increased risk of somnolence/sedation relative to placebo varied across: with olanzapine: odds ratio (OR) = 8.49 (3.97–16.55); risperidone: OR = 7.3 (4.63–11.19); aripiprazole: OR = 6.07 (2.79–12.22); and quetiapine: OR = 5.44 (2.91–9.26) (Cohen et al. 2012).
Furthermore, in open-label studies, rates of somnolence or fatigue were reported as 33% in aripiprazole, 44%–94% in olanzapine, 25%–80% in quetiapine and 29%–89% in risperidone (Zuddas et al. 2011). On the other hand, in a meta-analysis of youth with bipolar disorder, increased incidence rates for sedation/somnolence with SGAs compared to placebo did not differ across individual SGAs (Correll 2010), a finding that was replicated across several diagnoses in a later meta-analysis (Cohen et al. 2012).
Antipsychotic choice and dosing is thought to be key to the management or avoidance of side effects (Miller 2004; Correll et al. 2010). In our study, tranquilizing or activating effects could not clearly be related to dosing in all SGAs, probably due to the small sample sizes in all, but the risperidone treatment group. Indeed, in this group, higher dosing was consistently found in subjects with tranquilizing symptoms, not only during the early dose-finding period but also during the stable treatment period.
Moreover, dose associations were also found with regard to activating effects. However, lower risperidone doses were more likely present in patients complaining of restlessness or psychomotor activation, an observation that more likely reflects a lack of efficacy than a medication-related side effect, unless lower doses were due to attempts at minimizing akathisia.
Importantly, however, this observational study in which the clinician's treatment choice and management play a crucial role in determining efficacy and tolerability was not designed to study dose effects, which are much clearer seen in studies with predefined dose categories.
By contrast, one can benefit from the naturalistic design to identify more generalizable clinical predictors of side-effect profiles. Older subjects were relatively more sensitive to sedating effects, whereas younger subjects were more sensitive to activating effects. Moreover, subjects with lower baseline functioning (independent of age or clinical diagnosis), were generally more prone to experience either sedating or activating effects. This latter observation may reflect the higher neurobiological vulnerability in children with a more severe course of disease, such as seen in the form of microstructural cerebral changes such as seen in the form of microstructural cerebral changes in early psychoses (Fraguas et al. 2014).
Outpatient status was associated with higher rates of complaints about activating effects. Considering that a failure to decrease activating symptoms may reflect undertreatment, particularly since they were inversely dose related, these data suggest the need for closer monitoring of treatment efficacy in outpatients.
Paradoxically, despite the general use of lower doses in the treatment of youth with disruptive behavior-spectrum diagnoses, in whom aggression is a common treatment target, this diagnostic category, but not antipsychotic dosing per se, emerged as a predictor for reduced likelihood of psychomotor retardation. It is possible that the lack of a common dosing effect across SGA groups is related to the shortcoming of unified calculations of CPZEs, especially in the lower dose range (Leucht et al. 2015), which limits the option to detect dose–effect associations across different antipsychotic groups.
Interestingly, stimulant comedication was the strongest independent predictor of postbaseline psychomotor activation. Considering that psychomotor activation consistently declined during antipsychotic treatment, this result is suggestive of an attenuation of the antipsychotic-related sedating effects through stimulant comedication. Indeed, due to the contrasting effects of stimulants and antipsychotics on dopaminergic transmission, their interaction is to be expected. Although cotreatment of antipsychotics and stimulants occurs relatively frequently (Penzner et al. 2009), their interactions have not systematically been assessed. A recent claims database study found higher rates of healthcare resource utilization in subjects with antipsychotic-stimulant cotreatment (Sikirica et al. 2014).
It is concerning that, contrary to widespread conviction, tolerance of the sedating and activating symptoms could not be observed, at least not within the 3-month period. This observation is at least partially consistent with an earlier study of quasi-antipsychotic-naïve youth, which observed that even after 12 months of primarily SGA treatment, one-third of youth continued to experience UKU scale-rated somnolence/sedation (Merchán-Naranjo et al. 2012).
The findings of our study need to be interpreted within its limitations. These include the nonrandomized naturalistic design with inclusion of a heterogeneous group of psychiatric disorders. In this setting, the treatment allocation is vulnerable to a selection bias, in that clinicians may anticipate a side-effect profile of a specific SGA. For example, clinicians may be more likely to choose quetiapine as a first-line treatment in a restless subject, anticipating an effect on this secondary symptom. However, at least based on the patient perspective (i.e., reflected in TESS ratings), none of such baseline differences were present. Nevertheless, it is still possible that SGA groups were not fully matched in this regard.
On the other hand, the naturalistic design of this inception cohort study increases the generalizability of the findings, and the existing group differences between SGA groups were controlled in models searching to identify specific SGA-side effect associations.
Furthermore, sample sizes for aripiprazole, olanzapine, and quetiapine were around 40 only, which may increase the risk for a type II error. Moreover, baseline assessments occurred on average 2.7 days after the first SGA dose was received, potentially confounding the baseline measurement, even though patients were explicitly asked how they felt before the antipsychotic was started. Finally, we did not use sophisticated assessment tools for sleep and activity levels, such as sleep quality scales, polysomnography, or actigraphy, and did not assess the functional impact of the measured adverse effects, for example, on socialization, school functioning, or school grades. However, the low treatment discontinuation rates for the observed adverse effects, limited to sedation/drowsiness, suggest that the measured adverse effects were not functionally limiting in most cases.
Despite these limitations, strengths of this study include that we examined an antipsychotic-naïve sample in which carryover effects and channeling bias due to prior experiences with SGAs are excluded, that the four most commonly prescribed antipsychotics were compared within the same study, using the same methodology, and that we used a rating scale to proactively elicit the targeted patient-reported adverse effects. Moreover, different from most available studies, we did not only assess period incidence rates across the entire study period but also the trajectory of the adverse effects under investigation, accounting for potentially differential development of tolerance across SGAs.
Conclusions
Despite clinicians' efforts to optimize the benefit–side effect ratio, tranquilizing symptoms continue to be highly prevalent and persistent during continued periods of SGA treatment with a consecutive need to inform subjects at treatment initiation about this possibility. By contrast, activating symptoms remain restricted to a third of subjects at most.
Even though small differences across SGAs in the two compound side effect measures were noted, the predictors of single complaints, such as drowsiness or psychomotor retardation included clinical parameters, rather than specific SGAs or SGA doses. Individualized side effect management may improve with a more careful age-adapted dosing, as older youth proved to be more vulnerable to tranquilizing symptoms, while younger subjects were more sensitive to activating symptoms. Similarly, cautious treatment strategies may be needed for low-functioning youth who showed increased sensitivity to activating as well as sedating effects. Last, during outpatient management, closer collaboration between clinicians, patients, and caregivers may be needed, as activating symptoms improved insufficiently in this group.
Footnotes
Disclosures
Drs. Al-Dhaher, Kapoor, Saito, Krakower, David, and Mr. Ake have nothing to disclose. Dr. Kane has received honoraria for lectures and/or consulting from Alkermes, Bristol Myers Squibb, Eli Lilly, Forrest Labs, Forum, Genentech, IntraCellular Therapies, Janssen, Johnson and Johnson, Lundbeck, Merck, Novartis, Otsuka, Pfizer, Reviva, Roche, and Sunovion. He has received grant support from Genentech, Johnson and Johnson, and Otsuka. He is a shareholder of MedAvante and Vanguard Research Group. Dr. Correll has been a consultant and/or advisor to or has received honoraria from AbbVie, Actavis, Alkermes, Bristol-Myers Squibb, Eli Lilly, Genentech, Gerson Lehrman Group, IntraCellular Therapies, Janssen/J&J, Lundbeck, MedAvante, Medscape, Otsuka, Pfizer, ProPhase, Reviva, Roche, Sunovion, Supernus, and Takeda. He has received grant support from Bristol-Myers Squibb, Otsuka, and Takeda. Dr. Carbon has the same conflict of interest as Dr. Correll due to family relationship.