
Abstract
Objective:
To investigate the safety and pharmacokinetic profile of ascending doses of desvenlafaxine in children and adolescents with major depressive disorder. Assessment of the effect of desvenlafaxine on depression symptoms was exploratory.
Methods:
The 8-week, open-label study included an initial 3.5-day inpatient period followed by a 7.5-week outpatient period. Children (7–11 years) received a single desvenlafaxine dose of 10, 25, 50, or 100 mg on day 1; adolescents (12–17 years) received desvenlafaxine 25, 50, 100, or 200 mg/day. Plasma and urine samples were collected over the initial 72-hour inpatient period. Evaluations included treatment-emergent adverse events (TEAEs), physical examinations (including Tanner Staging), vital signs, laboratory assessments, 12-lead electrocardiogram, Columbia-Suicide Severity Rating Scale, and the Children's Depression Rating Scale-Revised (CDRS-R).
Results:
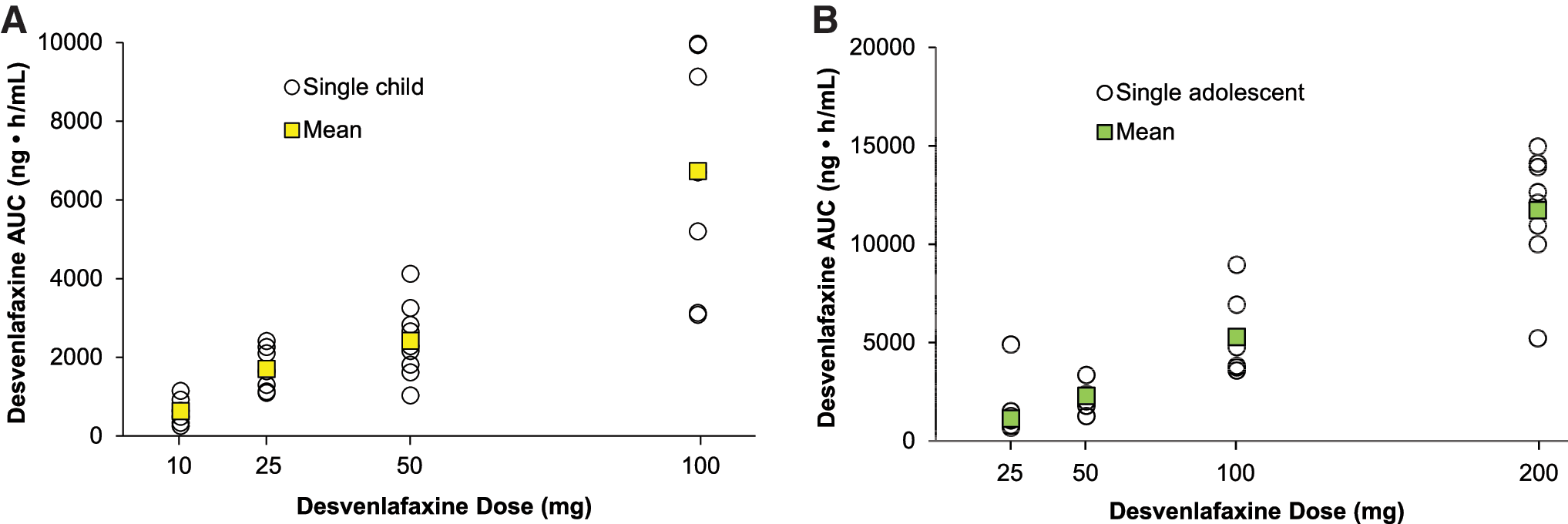
In all, 29 children and 30 adolescents took at least one dose of desvenlafaxine and were included in the safety population (children: 10 mg, n = 6; 25 mg, n = 7; 50 mg, n = 9; 100 mg, n = 7; adolescents: 25 mg, n = 7; 50 mg, n = 7; 100 mg, n = 8; 200 mg, n = 8). Total area under the drug concentration-time curve from 0 to infinity (AUC) appeared to increase linearly with increasing dose. Mean (standard deviation [SD]) AUC ranged from 628 (346) ng/mL (desvenlafaxine 10 mg) to 6732 (3031) ng/mL (100 mg) in children and from 1123 (361) ng/mL (25 mg) to 11,730 (3113) ng/mL (200 mg) in adolescents. During the combined inpatient and outpatient period, 16/29 (55%) children and 21/30 (70%) adolescents reported at least one TEAE. One serious adverse event (suicidal behavior) was reported. Mean (SD) change from baseline in CDRS-R total scores at week 8 was −19.00 (9.87) for children and −21.57 (11.50) for adolescents.
Conclusions:
Desvenlafaxine AUC values increased linearly with dose; body weight alone provided an adequate prediction for dose-normalized AUC. Desvenlafaxine was generally safe and well tolerated in children and adolescents for treatment up to 8 weeks.
Introduction
T
Clinical practice guidelines recommend treatment with proven antidepressant medications combined with psychological intervention such as cognitive behavioral therapy for adolescent patients with moderate to severe depression (Royal Australian College of General Practitioners 2005; Birmaher et al. 2007; Cheung et al. 2007; U.S. Preventive Services Task Force 2009), and in children (7 to 11 years) with moderate to severe depression, antidepressant treatment may be considered a first (Birmaher et al. 2007) or second-line treatment (National Collaborating Centre for Mental Health 2005; U.S. Preventive Services Task Force 2009). However, there is an unmet need for effective pharmacotherapies for children and adolescents with depression: to date, only two pharmacological treatments are approved by the U.S. Food and Drug Administration (FDA) for the treatment of pediatric patients with MDD (Food and Drug Administration 2014; Lexapro [package insert] 2014; Prozac [package insert] 2014). Guidelines stress the relative lack of evidence for antidepressant efficacy in pediatric populations, particularly in preadolescent children when compared to adult trials, and emphasize the need for additional research on the efficacy and safety of pharmacotherapies for children and adolescents with MDD (National Collaborating Centre for Mental Health 2005; Royal Australian College of General Practitioners 2005; Birmaher et al. 2007).
Pediatric clinical trials for antidepressant drugs can prove challenging. Enrollment of pediatric patients is limited due to the high percentage of patients who fail to meet inclusion and exclusion criteria for depression trials (Bliznak et al. 2013), and placebo response may be particularly high in children in multisite clinical trials for depression (Bridge et al. 2007, 2009). Pediatric safety and efficacy trials must therefore be designed to optimize the capability to identify safe and effective treatments. Dose selection is an important factor in study design, to avoid including doses below an efficacy threshold or above a tolerability range (Findling et al. 2006). Pharmacokinetic (PK) data are therefore critical for selecting appropriate study drug doses for pediatric efficacy and safety trials (Findling et al. 2006).
Desvenlafaxine (administered as desvenlafaxine succinate) is a serotonin–norepinephrine reuptake inhibitor (SNRI) approved by the U.S. FDA for the treatment of adult patients with MDD (Pristiq [package insert] 2015); it has not been approved for pediatric use. Desvenlafaxine is the major active metabolite of venlafaxine and is primarily eliminated through phase II glucuronidation and renal excretion, with minimal phase I hepatic metabolism by cytochrome P450 (CYP) 3A4 (DeMaio et al. 2011; Pristiq [package insert] 2015). Less than 5% of desvenlafaxine is excreted as the oxidative metabolite, N,O-didesmethylvenlafaxine (NODV) (Preskorn et al. 2008; Pristiq [package insert] 2015).
A pediatric program evaluating desvenlafaxine for the treatment of MDD in children (aged 7–11 years) and adolescents (aged 12–17 years) has been undertaken. As a first step, PK, safety, and tolerability of a range of desvenlafaxine doses were assessed in children (desvenlafaxine 10–100 mg/day) and adolescents (desvenlafaxine 25–200 mg/day) in an 8-week, open-label (OL) trial followed by a 6-month OL extension trial. The objectives of this initial lead in study were to investigate the safety and tolerability of ascending doses of desvenlafaxine in children and adolescents with MDD and characterize the PK profile of single ascending doses of desvenlafaxine in children and adolescents with MDD. The therapeutic effect of desvenlafaxine on symptoms of depression in children and adolescents with MDD was also evaluated in an exploratory manner. Safety and tolerability of the subset of patients who enrolled in the extension study have been reported (Findling et al. 2014); treatment with desvenlafaxine was generally safe and well tolerated in children and adolescents in that analysis. The results of the PK analysis and safety and effectiveness findings from the full population of patients enrolled in the lead-in study are reported in this study.
Patients and Methods
Study design
This phase 2a, multicenter, OL, safety, tolerability, and PK study of desvenlafaxine for the treatment of children and adolescents with MDD was conducted at six U.S. sites (Little Rock, AR; Cleveland, OH; Houston, TX; New York, NY; Wichita, KS; Terre Haute, IN) from February 2008 to November 2009. The study was conducted in accordance with the International Conference on Harmonization Guideline for Good Clinical Practice (International Conference on Harmonization 1998) and the ethical principles that have their origins in the Declaration of Helsinki. The study protocol and amendments received institutional review board or independent ethics committee approval. Parents provided witnessed, written informed consent; patients provided witnessed written assent.
The study included a 6- to 14-day screening period, an 8-week treatment period, a 0- to 2-week taper period, and a 1-week follow-up. The 8-week treatment period consisted of a 3.5-day inpatient PK study followed by 7.5-week outpatient treatment. Patients who completed the 8-week study and met entry criteria had the option of continuing in a 6-month OL continuation study. Combined safety data for the 8-week lead-in and the 6-month continuation studies were previously reported for patients who continued into the OL continuation study (40/59 patients enrolled in the 8-week study) (Findling et al. 2014). Safety data for all patients in the 8-week study safety population are reported in this study.
Desvenlafaxine once-daily doses of 10, 25, 50, and 100 mg were to be evaluated for PK in children; desvenlafaxine once-daily doses of 25, 50, 100, and 200 mg were to be evaluated for PK in adolescents. For each age stratum, the study drug was to be administered at only one dose level at a time for at least the first six patients per dose group. Administration at the next higher dose level within an age stratum was not to begin until the safety and tolerability of the preceding dose had been evaluated through 48 hours after administration and deemed acceptable by both the site investigators and the sponsor's medical monitor based on an evaluation of PK and safety data. The medical monitor role is accountable for medical oversight of the study, including clinical trial design, execution, safety review, interpretation of efficacy and safety data, and oversight of nonmedically qualified clinical colleagues. The medical monitor also addresses medical questions from investigative sites.
Patients were admitted to an inpatient unit on study day 1 for the PK assessment. On study day 1, each patient received a single oral dose of study drug at their assigned dose level after an overnight fast of at least 10 hours (Table 1). Plasma samples were collected predose and at postdose hours 0.5, 1, 2, 4, 6, 8, 12, 24, 36, 48, 60, and 72. Urine collections were completed predose and from 0 to 4, 4 to 8, 8 to 12, 12 to 24, 24 to 48, and 48 to 72 hours postdose. Patients received no additional study drug until the outpatient period, starting day 4.
No desvenlafaxine was administered on days 2 and 3.
—, indicates no desvenlafaxine dose administered.
Patients then received once-daily doses of study drug under parental supervision as appropriate starting on day 4 after collection of the 72-hour plasma sample. Patients' dose assignments were the same in the outpatient period as in the inpatient PK assessment period (children: 10, 25, 50, and 100 mg/day; adolescents: 25, 50, 100, and 200 mg/day), but the outpatient period included a titration phase. The lowest dose for each age group (children: 10 mg/day; adolescents: 25 mg/day) was not titrated; all other doses were titrated over the first 1 to 2 weeks of the outpatient period. Doses greater than 10 mg/day were tapered over 1 to 2 weeks upon completion of the study for those who did not continue into the OL extension study (Findling et al. 2014), or at early withdrawal. The titration and tapering schedules for each dose group are shown in Table 1.
Selection of patients
Study participants were child (7–11 years) and adolescent (12–17 years) outpatients with moderate to severe MDD without psychotic features. Diagnosis was based on Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM-IV-TR) (American Psychiatric Association 2000) criteria and assessed by the KIDDIE Schedule for Affective Disorders and Schizophrenia–Present and Lifetime Version (Kaufman et al. 1997) and confirmatory clinical interview. Entry criteria also included a Children's Depression Rating Scale-Revised (CDRS-R) (Poznanski et al. 1979) score greater than 40 and a Clinical Global Impressions Scale–Severity (CGI-S) (Guy 1976) score of 4 or greater at screening and baseline.
Patients were ineligible for the study if they had a medical condition known to interfere with the absorption or excretion of drugs; a clinically significant general medical illness within 90 days before screening or a mental disorder due to a general medical condition; a primary diagnosis of generalized anxiety disorder, panic disorder, social anxiety disorder, or attention-deficit hyperactivity disorder within 12 months before baseline; current (within 12 months before baseline) psychoactive substance abuse or dependence (including alcohol), posttraumatic stress disorder, obsessive-compulsive disorder, a diagnosis of bipolar disorder or psychotic disorder, or first-degree relative with bipolar disorder; clinically relevant abnormalities on screening physical examination, electrocardiogram (ECG), laboratory tests, or urine drug screen; or a clinically important personality disorder as assessed during the screening psychiatric evaluations.
Additional exclusion criteria included a history of suicide attempt or gesture; acute suicidality; screening or baseline CDRS-R question 13 (Suicidal Ideation) score of 5 or greater; 17-item Hamilton Depression Rating Scale (HAM-D17) (Hamilton 1960) Item 3 (Suicide) score of 3 or greater; “yes” determinations on Columbia Suicide-Severity Rating Scale (C-SSRS) (Posner et al. 2011) questions regarding Active Suicidal Ideation with Some Intent to Act Without Specific Plan, Active Suicidal Ideation with Specific Plan and Intent, or Actual Attempt; or if they have a first-degree relative who completed suicide. Patients were also excluded if they had a history or presence of anorexia or bulimia or their weight was below the 10th percentile for age based on the National Center for Health Statistics growth charts (Kuczmarski et al. 2002). Patients who were pregnant (positive serum beta-human chorionic gonadotropin test result during screening), breastfeeding, planning to become pregnant, and sexually active, who did not commit to the use of medically accepted forms of contraception during study participation, were excluded. Patients who received any investigational drugs or devices within 30 days before administration of the first dose of study drug were excluded. In addition, patients were given a placebo swallow test at screening, and were excluded if they were unable to swallow a tablet or tablets of size equal to the largest-sized desvenlafaxine tablet studied in their respective age stratum.
Prohibited treatments included ketoconazole and other strong CYP3A4 inhibitors (14 days before study day 1 until completion of the inpatient period); tobacco, caffeine-containing products, grapefruit or grapefruit-containing products, or alcohol (48 hours before study day 1 through completion of the inpatient period); venlafaxine (within 90 days before study day 1); antipsychotic drugs and fluoxetine (within 30 days before study day 1); sumatriptan, naratriptan, zolmitriptan, or drugs with a similar mechanism of action, tryptophan supplements, anxiolytics, antidepressants (other than fluoxetine or venlafaxine), and monoamine oxidase inhibitors (within 14 days before study day 1); and stimulants, atomoxetine, herbal products intended to treat anxiety, insomnia, or depression, other psychotropic drugs or substances, and sedative-hypnotic drugs (within 7 days before study day 1). Nonpsychopharmacologic drugs with possible psychotropic effects (such as antihistamines) were permitted only if a stable dose had been taken for at least 90 days (as-needed use of typical cold preparations was permitted).
Bioanalytical methodology
Plasma samples were analyzed for plasma desvenlafaxine concentrations, and urine samples were analyzed for desvenlafaxine, total desvenlafaxine (free plus glucuronide conjugate), NODV, and total NODV (free plus glucuronide conjugate) by Cetero Research (Houston, TX). Desvenlafaxine concentrations in plasma and desvenlafaxine and NODV concentrations in urine were measured using validated liquid chromatography/tandem mass spectrometry (LC/MS/MS) using nadolol (USP, Rockville, MD) as an internal standard. Quantitative determination of desvenlafaxine in plasma samples and desvenlafaxine and NODV in urine samples was carried out using an API 3000 LC/MS/MS system (Applied Biosystems, Foster City, CA) equipped with a PE Series 200 LC pump and autosampler, and a Betabasic CN analytical column (100 × 2.1 mm, 5 μm equipped with an in-line precolumn filter; Thermo Scientific, Waltham, MA) with a TurboIonspray® interface. Data collection and integration were performed using Applied Biosystems “Analyst” version 1.4.1 software. The slopes, intercepts, and correlation coefficients were determined by least-squares linear regression analysis using the ratios of analyte/internal standard peak areas of calibration curve standards in Watson LIMS version 6.4.0.02™ for Windows (Thermo Scientific). The weighting factor of 1/x2 (1/concentration2) was used in the calculation of linear regression line.
Blood sample analysis
For analysis of plasma samples, nine calibration curve standards and two blanks with internal standard and two blanks without internal standard were analyzed with each run. The accepted run had an r 2 value of 0.997087 or better for the calibration standard curves. The interday precision (% coefficient of variation [%CV]) for the calibration standard was 4.3% or better and the accuracy (%bias) ranged from −2.5% to 1.3%. At least two sets of quality control (QC) samples (concentrations: 5.000, 45.00, and 375.0 ng/mL) were assayed with each run. Interday precision (%CV) for the QC samples was 9.0% or better; accuracy (%bias) ranged from −2.4% to 3.0%. The precision and accuracy for the diluted high QC samples were 5.3% and −0.9%, respectively.
Plasma desvenlafaxine was quantitated using a protein precipitation extraction procedure. A total of 20.0 μL of deionized water was added to each 200 μL aliquot of standard and QC sample, and 20.0 μL of 50% methanol–water solution was added to each 200 μL aliquot of study sample; 0.50 mL of working internal standard solution (50.0 ng/mL) was added to each sample. After vortexing and centrifuging, 200 μL of the supernatant was transferred to an autoinjector vial. Following the addition of 1000 μL of dilution solution to each vial, 5.00 μL was injected onto the API 3000 LC/MS/MS system. The lower limit of quantitation (LLOQ) for desvenlafaxine in plasma samples was 2.000 ng/mL and the upper limit of quantitation (ULOQ) was 500.0 ng/mL. Data occurring before the first measurable concentration were set to zero; data reported as below the quantitation limit occurring between two measurable concentrations and after the last measurable concentration were set as missing.
Urine sample analysis
Urine samples were analyzed using nine calibration curve standards each for desvenlafaxine and NODV, and two blanks with internal standard and two blanks without internal standard were analyzed with each run. All accepted runs had r 2 values of 0.993794/0.991486 or better for desvenlafaxine (total)/desvenlafaxine (unconjugated) for the desvenlafaxine calibration standard curves and r 2 values of 0.994850/0.990279 or better for NODV (total)/NODV (unconjugated) for the NODV calibration standard curves. The interday precision (%CV) for total desvenlafaxine was 4.7% or better and the accuracy (%bias) ranged from −4.5% to 2.2%; interday precision (%CV) for unconjugated desvenlafaxine was 4.8% or better and the accuracy (%bias) ranged from −5.3% to 5.0%. For NODV (total), the interday precision (%CV) was 9.4% or better and the accuracy (%bias) ranged from −2.4% to 2.0%; interday precision (%CV) for unconjugated NODV was 6.2% or better and the accuracy (%bias) ranged from −3.6% to 4.8%. Two sets of QC samples (desvenlafaxine: 0.3000, 3.000, and 38.00 μg/mL; NODV: 0.1200, 1.200, and 15.20 μg/mL) were assayed with each run. Interday precision for the desvenlafaxine QC samples was 9.2% or better; accuracy ranged from −3.7% to −0.6%. For NODV QC samples, interday precision was 9.2% or better and accuracy ranged from −2.4% to 0.2%.
Total desvenlafaxine and NODV concentrations in urine were quantitated using an enzymatic hydrolysis and dilution extraction procedure. A total of 600 μL of working internal standard solution (500 ng/mL) and 30.0 μL of β-glucuronidase enzyme solution were added to each 50.0 μL aliquot of standard, QC sample, and study sample. After incubating in water for 18 hours at 37°C, the sample was vortexed and centrifuged. Following the transfer of 20.0 μL of the supernatant to an autoinjector vial, 1.30 mL of dilution solution was added and 5.00 μL was injected onto the LC/MS/MS system. Unconjugated desvenlafaxine and NODV in urine were quantitated using a dilution method, in which each 20.0 μL aliquot of standard, QC sample, and study sample was mixed with 0.20 mL of working internal standard solution (500 ng/mL). After vortexing and centrifuging, 20.0 μL of the supernatant was transferred to an autoinjector vial, 1.30 mL of dilution solution was added, and 5.00 μL was injected onto the LC/MS/MS system. The LLOQ for total and unconjugated desvenlafaxine in urine samples was 0.1000 μg/mL and the ULOQ was 50.00 μg/mL; the LLOQ for total and unconjugated NODV in urine samples was 0.04000 μg/mL and the ULOQ was 20.00 μg/mL. Data reported as below the quantifiable limit of the assay were set to zero.
PK analysis
The plasma and urine concentration-time data for each patient were analyzed by noncompartmental methods using WinNonlin v 5.1.1 software (Pharsight Corporation, Mountain View, CA). For plasma desvenlafaxine, peak concentration (Cmax) and time to Cmax (tmax) were read directly from the observed data. The terminal-phase disposition rate constant (λz) for individual concentration-time profiles was determined by the log-linear regression of at least three points judged to be in the terminal phase. Terminal-phase elimination half-life (t1/2) was calculated as t1/2 = ln2/λz. Total area under the drug concentration–time curve from time 0 to infinity (AUC) was estimated using the trapezoidal rule during the ascending portion of the curve and the log-trapezoidal rule during the descending portion of the curve. Apparent oral-dose clearance (Cl/F) was estimated as the quotient of dose to the AUC. Apparent volume of distribution for the terminal disposition phase (Vz/F) was calculated as a ratio of Cl/F to λz.
For urine desvenlafaxine and NODV, the amount excreted (Ae) into urine was calculated as Ae = concentration × urine volume, and renal clearance (ClR) was calculated as ClR = Ae/AUC. Percent excreted unchanged in urine (Ae%) was calculated for desvenlafaxine as follows: Ae (%) = (Ae/Dose) × 100; for NODV, Ae(%) was calculated as follows: Ae (%) = ([Ae × 1.056]/Dose) × 100, to account for differences in molecular weight between desvenlafaxine and NODV.
Safety assessments
Physician clinical assessments and administration of C-SSRS were completed at screening, baseline, and study day 4 (end of inpatient period), on study days 7, 21, 28, 42, 56, and during taper and follow-up. Physical examinations, including Tanner Staging (Tanner 1962), were completed at screening and study day 56. Vital signs measurements were made at screening, baseline, days 1, 2, 3, 4, 7, 21, 28, 42, 56, and during taper and follow-up, and 12-lead ECG recordings were made at screening, baseline, and study days 1 and 56. Laboratory assessments were conducted at screening and study days 28 and 56. Adverse events (AEs) were collected throughout the study period and categorized based on the Medical Dictionary for Regulatory Activities.
Therapeutic assessments
The evaluation of treatment effects on depression symptoms was exploratory in this OL study. Assessments included CDRS-R, HAM-D17, CGI-S, and Clinical Global Impressions Scale–Improvement (CGI-I) (Guy 1976). The CDRS-R and CGI-S were administered at screening, baseline, and study days 4, 7, 14, 21, 28, 42, and 56. The HAM-D17 was administered at screening, baseline, and study days 4, 14, 28, and 56. The CGI-I was administered at study days 4, 7, 14, 21, 28, 42, and 56.
Sample size
All efficacy analyses were descriptive in nature; therefore, sample size was based on PK assessment needs. For a preliminary assessment of desvenlafaxine PK in pediatric patients, a sample size of approximately six patients for each age group (i.e., children and adolescents) per dose group combination was considered adequate.
Statistical methods
The PK analysis population was defined as all patients who had available concentration data, and whose concentration data were properly identified with respect to dosing and sampling times. The safety population was defined as all patients assigned to treatment who had taken at least one dose of study drug. Therapeutic effects were assessed for the intent-to-treat population (all patients who took at least one dose of study drug and had baseline and at least one postbaseline CDRS-R assessment).
Plasma and urine PK parameters were summarized with descriptive statistics. A preliminary population PK analysis was conducted to examine the relationship between AUC and body weight in children and adolescents. In that analysis, dose-normalized AUC (AUC/D) was regressed against body weight, using the following power equation: Y = A × Wb, where “Y” was AUC/D, “W” body weight, “A” the coefficient, and “b” the exponent. The analysis was performed using data from children alone, adolescents alone, and children and adolescents combined.
Mean changes in vital signs, ECG parameters, and laboratory evaluations were analyzed using paired t-tests. The proportions of patients with at least one potentially clinically important (PCI) value, based on FDA- or sponsor-defined criteria, for vital signs, ECGs, and laboratory evaluations over the inpatient treatment period were summarized across age–dose cohorts, and treatment-emergent adverse events (TEAEs) were tabulated by age–dose cohort. Tanner Staging and C-SSRS responses were summarized for each age–dose cohort and the overall analysis population.
Assessment of treatment effects on depression symptoms was exploratory only. Mean change from baseline in CDRS-R and HAM-D17 total scores was summarized at each time point for the overall population, for children and adolescents separately, and for each age–dose cohort. For CGI-S and CGI-I, the proportions of patients giving each categorical response were summarized at each time point overall, for children and adolescents separately, and by age–dose cohort. The last observation carried forward (LOCF) approach was used to evaluate missing data.
Results
Patients
A total of 79 patients were screened; 59 patients (29 children, 30 adolescents) were sequentially assigned to treatment (Fig. 1). All 59 took at least one dose of study drug and were included in the safety population. As previously reported (Findling et al. 2014), 2/29 (6.9%) children discontinued before completing 8 weeks of treatment; one child assigned to desvenlafaxine 50 mg/day was discontinued by investigator request (patient refused to cooperate with blood draw), and one assigned to desvenlafaxine 100 mg/day was discontinued due to a protocol violation (noncompliance taking study drug). A total of 6/30 (20.0%) adolescents discontinued before completing 8 weeks of treatment. Four adolescents discontinued due to AEs (suicidal behavior [25 mg/day], affect lability [25 mg/day], infectious mononucleosis [200 mg/day], upper abdominal pain and muscle spasms, [200 mg/day]), and two discontinued due to protocol violations (did not return due to death in family [100 mg/day], noncompliance [200 mg/day]).
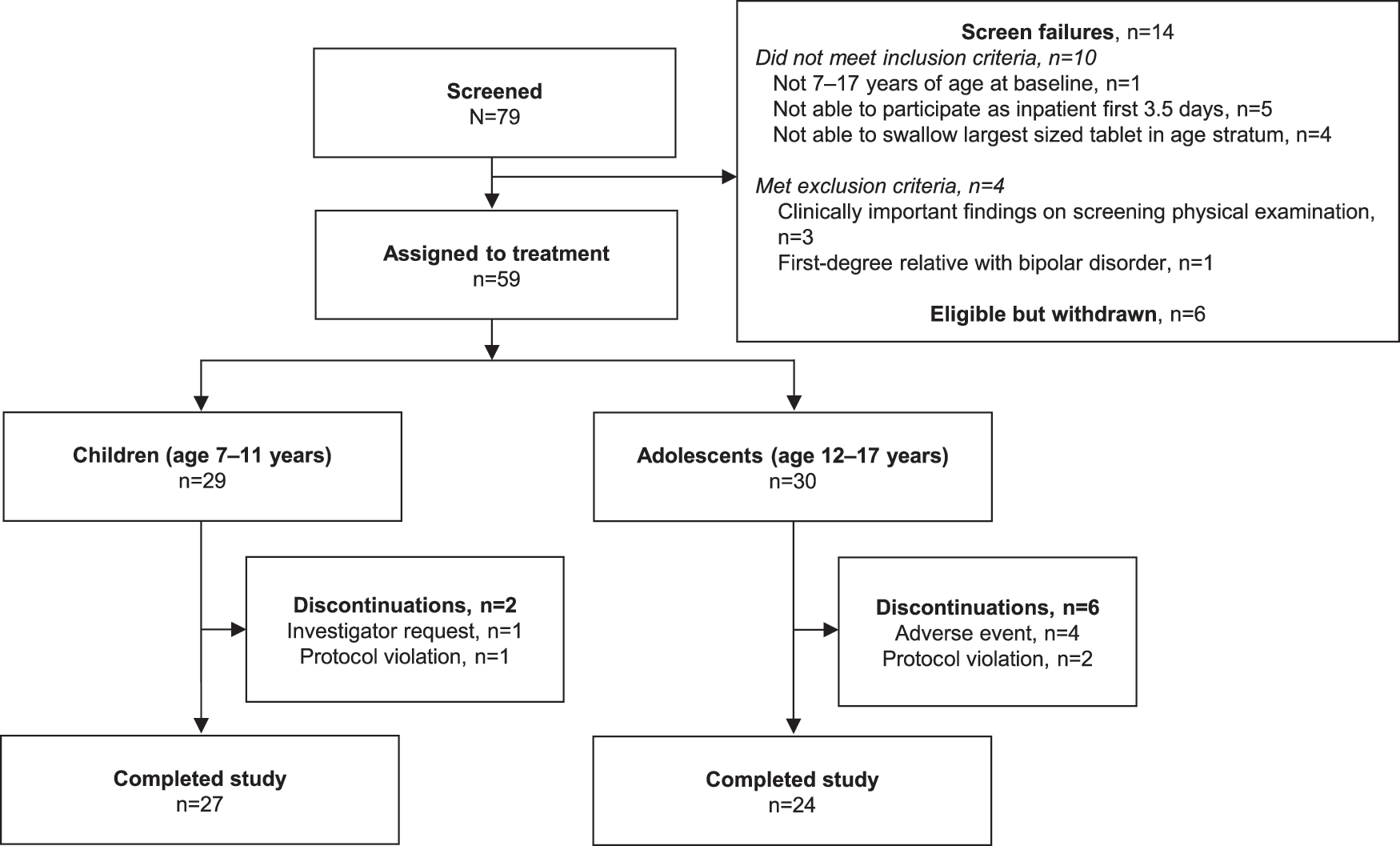
Study flowchart.
Demographic and baseline characteristics were similar across treatment groups (Table 2). Children enrolled in the study ranged in age from 7 to 11 years (mean, 9.6 years) and adolescents ranged from 12 to 17 years (mean 14.1). A total of 14/29 children and 15/30 adolescents in the safety population were female. The mean duration of the current episode of MDD at the time of screening was 7.2 months (range, 1.1 to 24.0 months) for children and 9.0 months (range, 2.0 to 46.6 months) for adolescents. Mean CDRS-R score at baseline was 51.2 (range, 42 to 66) for children and 57.0 (range, 43 to 78) for adolescents. Based on a CGI-S score of 4, 96.6% of children and 90.0% of adolescents were considered moderately ill at baseline.
BMI, body mass index; SD, standard deviation.
Pharmacokinetics
PK data were available for 29 children (10 mg, n = 6; 25 mg, n = 7; 50 mg, n = 9; 100 mg, n = 7) and 28 adolescents (25 mg, n = 7; 50 mg, n = 7; 100 mg, n = 6; 200 mg, n = 8). Of those, two children in the 50-mg group and one adolescent in the 200-mg group had missing urine samples and were not included in the summary of urinary excretion data. In addition, the sponsor made a decision to exclude two adolescents in the 100-mg group from all PK analyses due to errors in identification of their plasma and urine samples at the site.
The FDA issued a letter on August 3, 2011 informing Pfizer of suspected data falsification of bioanalytical data generated by Cetero Research during a time period that included the analysis of samples from this study. Pfizer examined data study by study in a thorough audit of clinical studies for desvenlafaxine, which included bioanalytical data generated by Cetero during the period from June 2005 to May 2009. In instances where discrepancies were noted, clinical study reports were modified based on removal of suspect data from the database and reanalysis of all databases from which data were removed. The database for the current PK analysis was revised as follows: unconjugated desvenlafaxine and NODV urine concentration values for one patient in the 25 mg/child group, two patients in the 100 mg/adolescent group, and all patients in the 50 mg/child group were removed from the analyses. Descriptive statistics were recalculated for the 25 mg/child and 100 mg/adolescent groups, and the clinical study report was revised to reflect all changes related to the excluded data points.
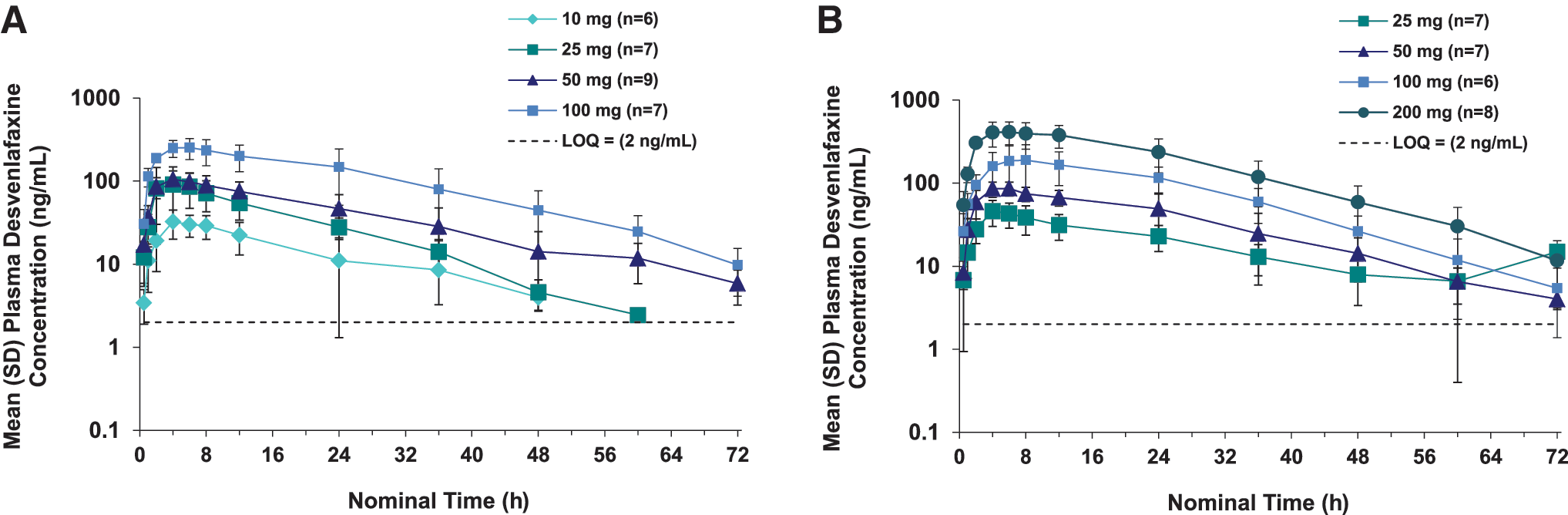
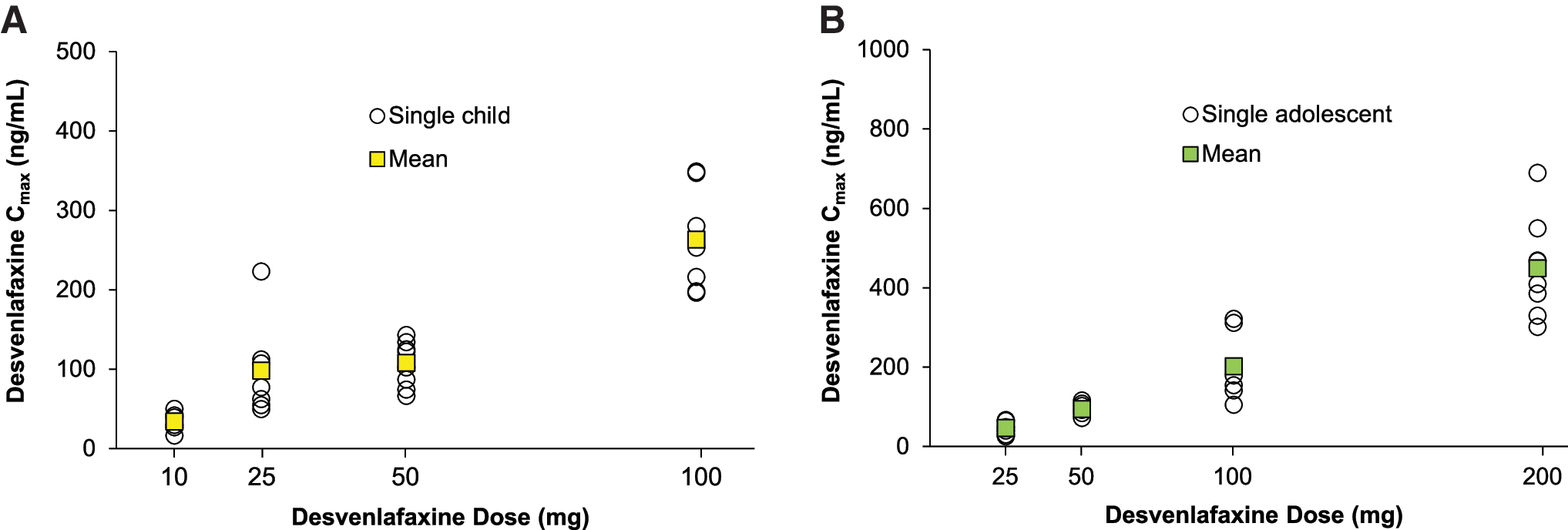
Summary statistics for plasma desvenlafaxine PK parameters are listed in Table 3; plasma desvenlafaxine concentration-time profiles are shown for children and adolescents in Figure 2A and B, respectively. In both children and adolescents, Cmax (Fig. 3) and AUC (Fig. 4) appeared to increase linearly with increasing dose. For children, mean (standard deviation [SD]) Cmax ranged from 33.9 (12.1) ng/mL for the desvenlafaxine 10 mg dose to 263 (66) ng/mL for the 100 mg dose, and mean AUC ranged from 628 (346) ng/mL to 6732 (3031) ng/mL. For adolescents, mean (SD) Cmax ranged from 46.1 (15.9) ng/mL for the desvenlafaxine 25 mg dose to 449 (126) ng/mL for the 200 mg dose; mean AUC ranged from 1123 (361) ng/mL to 11,730 (3113) ng/mL.
AUC, total area under the plasma concentration versus time curve; Cmax, peak plasma concentration; Cl/F, clearance; tmax, time of peak concentration; t1/2, mean terminal elimination half-life; Vz/F, apparent volume of distribution.
Dose-normalized AUC was adequately predicted by body weight alone (without age). The effect of body weight on dose-normalized AUC could be described by an exponential equation for each age group. Dose-normalized AUC could be described in children as follows: AUC/D = 3180.6 × W(−0.6022). In adolescents, AUC/D = 599.46 × W(−1.1054), and for the total PK population (children + adolescents), AUC/D = 476.83 × W(−0.5673). Mean CL/F was generally higher in children than in adolescents, with large variability within groups and no evidence of increases or decreases in CL/F with increases in dose (Table 3). CL/F was 54% higher in children compared with adolescents at the 25-mg dose, 22% higher at the 50-mg dose, and 56% higher at the 100-mg dose.
Table 4 lists the mean desvenlafaxine dose fraction excreted as total and unconjugated desvenlafaxine and total and unconjugated NODV for children and adolescents by dose group. Mean renal clearance of unconjugated desvenlafaxine in children was 0.125, 0.179, and 0.145 L/h/kg for the 10, 25, and 100 mg doses, respectively. In adolescents, mean renal clearance of unconjugated desvenlafaxine was 0.108, 0.173, 0.127, and 0.125 L/h/kg for the 25, 50, 100, and 200 mg doses, respectively.
For unconjugated desvenlafaxine and unconjugated NODV, n = 6 due to redacted data.
For unconjugated desvenlafaxine and unconjugated NODV, n = 4 due to redacted data.
For total desvenlafaxine, n = 6 due to a value above the quantitation limit in one patient.
NODV, N,O-didesmethylvenlafaxine; NR, values not reported, analytical results redacted from the analytical report; SD, standard deviation.
Safety and tolerability
Safety data for the 40 patients who continued into the OL continuation study were previously reported for both the 8-week lead-in and the 6-month continuation studies (Findling et al. 2014). Safety data for all 59 patients in the 8-week study safety population (children, n = 29; adolescents, n = 30) are reported here. One adolescent patient assigned to the desvenlafaxine 100 mg/day group received 50 mg/day during the outpatient period due to a dispensing error; data from this patient are reported in the 50 mg/day group.
In all, 16/29 (55%) children and 21/30 (70%) adolescents reported one or more TEAE during the combined inpatient and outpatient period. TEAEs reported by more than one patient in either age group are listed for the combined inpatient and outpatient period in Table 5. No patient died during the study. One serious AE was reported: an adolescent assigned to desvenlafaxine 25 mg/day experienced a serious AE of suicidal behavior on study day 30, 2 days after the patient's last recorded dose of study drug (the patient was noncompliant with study drug dosing). The patient was formally discontinued from the study drug and was hospitalized with parental agreement. The patient was discharged after 6 days, and the patient's parent chose not to return to the study site for a follow-up visit after the patient was discharged. C-SSRS scores for the patient indicated no suicidal thoughts, ideation, or attempts before study entry, or while on study through the patient's final on-study evaluation, which took place before the serious AE; no C-SSRS evaluation was completed for the patient after the serious AE.
Percentage based on females.
TEAE, treatment-emergent adverse event.
No lifetime history of suicide-related thoughts or behaviors was reported for any child based on the C-SSRS administered at the screening visit; four adolescents (desvenlafaxine 25 mg/day, one patient; desvenlafaxine 50 mg/day, one patient; desvenlafaxine 100 mg/day, two patients) had suicidal thoughts and three adolescents (desvenlafaxine 25 mg/day, one patient; desvenlafaxine 100 mg/day, two patients) had self-injurious behavior with no suicidal intent before study entry (lifetime). Based on C-SSRS results at the baseline study visit, two adolescents and no children had suicidal ideation during the screening period. One adolescent (100 mg/day) who had suicidal ideation before study entry (based on baseline C-SSRS results) had suicidal ideation while on study at two postbaseline study visits (study days 4 and 7). No other suicide-related thoughts or behaviors were reported by the C-SSRS assessments at the baseline or postbaseline study visits.
PCI laboratory results were observed for 13/27 (48%) children and 14/29 (48%) adolescents at some time during the therapy period. One or more laboratory values for eight children and 10 adolescents were considered clinically important by the medical monitor; eight children and nine adolescents had clinically important positive urine protein albumin dipstick test results. Among the children, five findings were trace urine protein albumin (5–20 mg/dL) at week 8 (desvenlafaxine 10 mg/day, one patient; 25 mg/day, two patients; 100 mg/day, two patients), with creatinine levels ranging from 0.53 to 0.77 mg/dL. The other three children (desvenlafaxine 25 mg/day, one patient; 50 mg/day, one patient; 100 mg/day, one patient) had 1+ urine protein albumin results (∼30 mg/dL) at week 8 (creatinine, 0.52 to 0.58 mg/dL); two of those three had trace urine protein albumin results at baseline. Adolescents with positive urine protein albumin test results included five patients with trace urine protein albumin results at week 8 or later (desvenlafaxine 25 mg/day, one patient; 100 mg/day, one patient; 200 mg/day, three patients; creatinine, 0.64 to 1.00 mg/dL), three with 1+ urine protein albumin results at week 8 (one patient each in the desvenlafaxine 25, 100, and 200 mg/day groups; creatinine, 0.57 to 0.80 mg/dL), and one with a 2+ urine protein result (∼100 mg/dL) (desvenlafaxine 50 mg/day; creatinine, 0.70 mg/dL). One adolescent with a 1+ urine protein albumin result (desvenlafaxine 25 mg/day) had clinically important low hematocrit at baseline and throughout the study (0.35–0.36 L/L). One adolescent had elevated AST/SGOT at week 4 (180 mU/mL; desvenlafaxine 100 mg/day) with results in the normal range at all other time points (baseline, 38 mU/mL; week 6, 24 mU/mL; week 8, 28 mU/mL). No symptoms were associated with the elevated AST/SGOT finding, no AEs were reported, and no treatments were provided. No other PCI laboratory results were considered to be clinically significant upon medical monitor review
A total of 24/29 (83%) children and 19/30 (63%) adolescents had at least 1 PCI vital sign result at some time during the therapy period. Four children (0 adolescents) had clinically important vital sign findings upon review by the medical monitor; all four had clinically important elevated systolic blood pressure measurements at three or more consecutive outpatient visits, based on PCI thresholds for their respective age and height. A 9-year-old male (124 cm in height) assigned to desvenlafaxine 25 mg/day had elevated supine systolic blood pressure measurements (>116 mm Hg) at each outpatient visit (baseline 89.3 mm Hg); his final poststudy measurement was 118 mm Hg. An 8-year-old male (134 cm) assigned to desvenlafaxine 50 mg/day had elevated supine systolic blood pressure measurements (>120 mm Hg) at inpatient day 4 and at outpatient visits starting at week 2 (baseline 108.7 mm Hg), with a final poststudy measurement of 124 mm Hg. Elevated blood supine systolic pressure measurements (>125 mm Hg) were observed intermittently during the inpatient period and at outpatient visits starting at week 2 in an 11-year-old male (149 cm) assigned to desvenlafaxine 50 mg/day (baseline 108 mm Hg). His final poststudy measurement was 140 mm Hg. An 11-year-old male (154 cm) assigned to desvenlafaxine 50 mg/day (baseline 120 mm Hg) had intermittent elevated supine systolic blood pressure measurements (>125 mm Hg) during the inpatient period and at all outpatient visits starting at week 2. He had no final poststudy measurement; at week 8, his supine systolic blood pressure was 136 mm Hg. No AEs related to blood pressure elevations were reported for these patients and none of the four patients received treatment with nonstudy medication. All other PCI vital signs findings were not considered to be clinically significant upon medical monitor review. Statistically significant changes from baseline in blood pressure and pulse rate were observed at final evaluation for some dose groups among children and adolescents (Table 6).
P < 0.05 versus baseline.
P < 0.001 versus baseline.
P < 0.01 versus baseline.
BP, blood pressure.
PCI ECG results were found in 16/29 (55%) children and 16/30 (53%) adolescents at some time during the therapy period. Upon medical monitor review, no PCI ECG findings were considered to be clinically significant.
Tanner Staging showed no change from baseline at week 8 (LOCF) for female children. In male children, a shift of two categories (from 1 to 3) for pubic hair was noted in one patient; a shift from 1 to 3 was also observed for testes in the same patient. Two adolescent girls shifted from category 4 to 5 for breasts and pubic hair. In adolescent boys, a shift in one patient from category 4 to 5 was observed for penis, pubic hair, and testes. Mean change in height from baseline to week 8 (LOCF) ranged from −0.07 to 0.33 cm across desvenlafaxine dose groups for children, with no statistically significant changes from baseline. Mean weight increased significantly from baseline to final evaluation in children treated with desvenlafaxine 25 mg/day (+0.62 kg; p < 0.05) or desvenlafaxine 50 mg/day (+1.34 kg; p < 0.05). For adolescents, mean change in height from baseline at week 8 (LOCF) ranged from −0.03 cm to 0.87 cm across desvenlafaxine dose groups. Mean weight change from baseline to final evaluation in adolescents ranged from −0.89 kg for the desvenlafaxine 200 mg/day group to +0.50 kg for the desvenlafaxine 25 mg/day group; none of the groups had a statistically significant mean weight change from baseline.
Therapeutic assessments
Mean (SD) change from baseline in CDRS-R total scores at week 8 (LOCF) was −19.00 (9.87) for children and −21.57 (11.50) for adolescents (baseline mean [SD]: children, 51.2 [6.66]; adolescents, 57.0 [9.71]). Mean (SD) change from baseline in HAM-D17 total scores at week 8 (LOCF) was −11.21 (5.73) and −11.80 (5.97) for children and adolescents, respectively (baseline mean [SD]: children, 16.7 [4.79]; adolescents, 19.4 [5.92]). At week 8 (LOCF), 72.4% of children and 70.0% of adolescents were CGI-I responders (CGI-I score of 1 [very much improved] or 2 [much improved]).
Discussion
In this study, single dose desvenlafaxine AUC values in children (desvenlafaxine 10 to 100 mg/day) and adolescents (desvenlafaxine 25 to 200 mg/day) were reasonably predicted based solely on body weight. The effect of body weight on dose-normalized AUC could be described by an exponential equation for each age group, and body weight alone provided an adequate prediction for AUC in a population PK analysis. Linear increases in Cmax and AUC were observed for increasing desvenlafaxine doses in children and adolescents. Mean CL/F values were higher in children (0.441–0.540 L/h/kg) compared with adolescents (0.282–0.441 L/h/kg). Mean CL/F values in adolescents were comparable to CL/F values for healthy adults (0.267–0.336 L/h/kg, desvenlafaxine 100 mg) (Nichols et al. 2011; Baird-Bellaire et al. 2013).
Desvenlafaxine was generally safe and well tolerated over the dose range assessed in children and adolescents in this study. Tolerability appeared to be dose related; overall TEAE rates were highest for the highest dose groups in each age cohort. The TEAEs reported by children and adolescents were consistent with the most common AEs reported in clinical trials for the treatment of MDD in adults (Clayton et al. 2009). Those that occurred at the highest rates (>10%) in children (headache, upper abdominal pain, vomiting, cough, and oropharyngeal pain) and adolescents (somnolence, nausea, headache, and upper abdominal pain) are also generally among the most common TEAEs reported in pediatric clinical trials of the SNRI duloxetine (Atkinson et al. 2014; Emslie et al. 2014) and venlafaxine (Emslie et al. 2007), and the selective serotonin reuptake inhibitor (SSRI) fluoxetine (March et al. 2004; Emslie et al. 2006; Emslie et al. 2014). Results of the C-SSRS indicated no signal for increased suicidal ideation or behaviors during the study, although there was one SAE of suicidal behavior by an adolescent. That event was not captured by the C-SSRS because it occurred after the patient's final evaluation.
There has been a history of concern about cardiovascular effects of SNRIs, specifically related to their noradrenergic component, introduced by early observations of elevated blood pressure in patients treated with venlafaxine (Feighner 1995). An increased occurrence of sustained elevation in diastolic blood pressure (≥90 mm Hg) was associated with venlafaxine at high doses (>300 mg/day) only (Feighner 1995; Thase 1998); however, the U.S. FDA prescribing information recommends regular or periodic blood pressure monitoring throughout treatment for adult patients treated with SNRIs (Fetzima [package insert] 2014; Cymbalta pacakge insert 2015; Effexor [package insert] 2015; Pristiq [package insert] 2015). Desvenlafaxine, which has a binding affinity at the human norepinephrine transporter in the same range as venlafaxine (Shelton 2009), was associated with mean changes from baseline in diastolic and systolic blood pressure of ∼2 mm Hg in an integrated analysis of 1834 adult MDD patients receiving desvenlafaxine doses ranging from 50 to 400 mg/day (Clayton et al. 2009). In the pediatric population in this study, statistically significant changes from baseline were observed in some dose groups at final evaluation, although mean changes from baseline were increases for some dose groups and decreases for others. No clear dose relationship was observed. Given that no adjustment was used for multiple comparisons and the study lacked a placebo control, the statistical significance of mean blood pressure changes in some dose groups must be interpreted with caution. The occurrence of clinically important sustained elevated systolic blood pressure in 4/29 children in this trial, however, underscores the recommendation for regular blood pressure monitoring if patients are being treated with SNRIs.
Assessment of therapeutic effects of desvenlafaxine on depression symptoms was exploratory in this OL trial. However, the change from baseline in CDRS-R and HAM-D17 total scores was consistent with a reduction in the severity of symptoms of depression, and the majority of both children and adolescents (≥70%) were either much improved or very much improved based on CGI-I scores at final evaluation. It will be critical, however, to assess the antidepressant efficacy of desvenlafaxine versus placebo before drawing conclusions regarding its effects on symptoms of depression in children and adolescents. Although meta-analyses have demonstrated an advantage for some SSRIs compared with placebo for treating MDD in pediatric patients, individual pediatric trials often fail to show a difference between the active drug and placebo (Tsapakis et al. 2008; Ma et al. 2014). An elevated placebo response particularly in children has been reported in pediatric trials for antidepressant efficacy (Cheung et al. 2006; Findling et al. 2006; Bridge et al. 2009). Based on the safety and tolerability results, together with the exploratory findings for therapeutic response, definitive phase 3 studies of desvenlafaxine in pediatric populations are warranted, and two placebo-controlled studies assessing the safety and efficacy of desvenlafaxine employing doses ranging from 20 to 50 mg/day are currently ongoing (
Limitations
This study was limited by the small number of patients per treatment group, its OL design, its brevity, and absence of a placebo control. Although the most common TEAEs in this study were consistent with those reported in adult MDD clinical trials, possible serious, but rare events may not have been detected in this small pediatric sample. Desvenlafaxine exposure in a larger patient population will be necessary to more thoroughly assess the safety and tolerability of desvenlafaxine in pediatric patients. Furthermore, the population of patients enrolled in this study was generally free of comorbid physical and psychiatric conditions, and therefore may not be representative of the pediatric patients treated in clinical practice. Urine PK results were limited by the redaction of suspect data.
Conclusions and Clinical Significance
For single doses of desvenlafaxine administered to children and adolescents, AUC values were reasonably predicted based solely on body weight. Linear increases in Cmax and AUC were observed for increasing desvenlafaxine doses in both children (10–100 mg/day) and adolescents (25–200 mg/day). CL/F was higher in children than in adolescents, but with considerable overlap in values. Clearance values for adolescents were similar to values observed for adults. Desvenlafaxine was generally safe and well tolerated in children and adolescents treated for up to 8 weeks in this study, and results of an exploratory assessment suggest that desvenlafaxine may reduce the symptoms of depression in children and adolescents with MDD. These results were informative for the design of definitive phase 3 efficacy and safety studies of desvenlafaxine that are currently underway in pediatric patients with MDD.
Note on PK Analysis Results
The FDA notified pharmaceutical companies of objectionable conditions at Cetero's Houston, TX, bioanalytical facility following several inspections. Plasma concentration samples for determination of desvenlafaxine and urine concentrations for concentration of desvenlafaxine and NODV were assayed at Cetero during a time period when the FDA requested an independent third-party audit to confirm the validity of the data. Pfizer conducted a thorough examination and verification of the bioanalytical data generated by Cetero's Houston, TX, bioanalytical facility for this study and has determined that concentration data obtained for this study are acceptable and accurate. The independent third-party audit has also been conducted as requested by the FDA. Five plasma samples for the assay of desvenlafaxine from five patients were found to be of potential risk. Eighteen urine samples from five patients for either desvenlafaxine or NODV were found to be at potential risk. Exclusion of these plasma and urine samples from descriptive statistics resulted in only minor changes to mean plasma and total urine concentration values and PK parameters.
Footnotes
Disclosures
Dr. Findling receives or has received research support, acted as a consultant and/or served on a speaker's bureau for Alcobra, American Academy of Child & Adolescent Psychiatry, American Physician Institute, American Psychiatric Press, AstraZeneca, Bracket, Bristol-Myers Squibb, CogCubed, Cognition Group, Coronado Biosciences, Dana Foundation, Elsevier, Forest, GlaxoSmithKline, Guilford Press, Johns Hopkins University Press, Johnson & Johnson, Jubilant Clinsys, KemPharm, Lilly, Lundbeck, Merck, NIH, Neurim, Novartis, Noven, Otsuka, Oxford University Press, Pfizer, Physicians Postgraduate Press, Purdue, Rhodes Pharmaceuticals, Roche, Sage, Shire, Sunovion, Supernus Pharmaceuticals, Transcept Pharmaceuticals, Validus, and WebMD. J.G., K.A.T., S.A.R., D.C., L.Y., and A.I.N. are Pfizer employees. J.G., K.A.T., and A.I.N. were employees of Pfizer at the time this study was conducted.