
Abstract
Objective:
The purpose of this study was to assess the long-term safety and tolerability of edivoxetine, a selective norepinephrine reuptake inhibitor, which was being developed as monotherapy in pediatric attention-deficit/hyperactivity disorder (ADHD).
Methods:
This was an open-label study of edivoxetine once daily dosing (0.1–0.3 mg/kg) as treatment for ADHD in children (6–11 years) and adolescents (12–17 years) to assess safety for up to 5 years. The safety assessments included the incidence of adverse events, vital signs, electrocardiograms, laboratory tests, percentile changes in weight, height, and body mass index, and Tanner staging. Efficacy of treatment with edivoxetine was also assessed using the Attention-Deficit/Hyperactivity Disorder Rating Scale-Version IV-Parent Reported: Investigator Scored (ADHDRS-IV) and Clinical Global Impressions-ADHD-Severity (CGI-ADHD-S).
Results:
A total of 267 children and adolescents were enrolled and 20 completed the 5-year study. Most of the participants were male (70.4%) and white (67.4%), and the mean age was 11.6 years. Two hundred three participants (76.9%; N = 264) experienced at least one adverse event. Treatment-emergent adverse events reported in >10% of participants were headache, vomiting, nausea, and upper respiratory tract infection. Serious adverse events (SAEs) were reported by seven participants (2.7%) during study treatment periods, and one participant was diagnosed with suspect epilepsy during the follow-up period after discontinuation of edivoxetine.
Conclusion:
Long-term open-label treatment with edivoxetine as monotherapy in children and adolescents with ADHD revealed a safety profile that was consistent with its pharmacological effects on norepinephrine transmission and with that reported in short-term studies of edivoxetine. The study was terminated early due to slow enrollment and the very low number of 5-year completers. Lilly is not proceeding with further development of edivoxetine, as announced in 2013.
Introduction
A
Because ADHD is often treated with medication for prolonged periods, there is a need to examine the tolerability and safety profiles of these treatments over time in children and adolescents. Of the nonstimulants, atomoxetine has been studied the most extensively. Donnelly et al. (2009) pooled data from 13 double-blind, placebo-controlled trials and 3 open-label extension studies of atomoxetine. In total, 714 patients were treated with atomoxetine for ≥3 years (mean follow-up 4.8 years [SD 1.1 years]), including a subset of 508 treated for ≥4 years (mean follow-up 5.3 years [SD 0.8 years]). No new or unexpected adverse events were observed during long-term exposure compared with acute treatment. In addition, there were no clinically significant long-term effects seen on growth rate, vital signs, or electrocardiographic parameters, and <2% of participants showed potentially clinically significant hepatic changes (Donnelly et al. 2009).
Edivoxetine hydrochloride (hereafter referred to as edivoxetine) is a highly selective and potent norepinephrine reuptake inhibitor that was being developed as a potential treatment for ADHD. Edivoxetine appears to depend less on the CYP2D6 enzyme for metabolism than atomoxetine (Michelson et al. 2007), and may have less exposure variability in patients with diverse CYP2D6 polymorphisms (Kielbasa et al. 2012). Edivoxetine demonstrated efficacy and safety in children and adolescents with ADHD in a small open-label trial of up to 1 year (Jin et al. 2013) and in a fixed-dose, randomized, double-blind, 8-week study (Lin et al. 2014). The purpose of this study was to assess long-term (up to 5 years) safety and tolerability of edivoxetine as monotherapy in pediatric ADHD.
Methods
Overview of study
This was an open-label, long-term safety and tolerability study of edivoxetine as monotherapy in children and adolescents with ADHD (
Enrollment began in September 2009 and the last participant completed the study in July 2015. Based on the enrollment information from a long-term pediatric study with atomoxetine (Spencer et al. 2007), the sample size for this study was estimated to be ∼1200 to have ∼100 participants reaching a 5-year exposure of edivoxetine. However, the current study was terminated early due to slow enrollment and the very low number of 5-year completers.
Participant selection
Children and adolescents who had completed a previous pediatric edivoxetine study, whether or not they were assigned to an edivoxetine treatment arm, could rollover into this study if they met the following inclusion criteria at the time of entry into their previous edivoxetine study: were aged at least 6 and <18 years at the time of informed consent; met Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM-IV-TR) diagnostic criteria for ADHD (inattentive, hyperactive/impulsive, or combined subtypes) (APA 2000), based on an interview by an experienced clinician and confirmed using the Kiddie Schedule of Affective Disorders and Schizophrenia for School-Aged Children Present and Lifetime (K-SADS-PL) Version (Kaufman et al. 1997); had a total score of at least 1.5 standard deviations (SDs) above the age and gender norm on the Attention-Deficit/Hyperactivity Disorder Rating Scale-IV-Parent Version: Investigator Administered and Scored (ADHDRS-IV-Parent:Inv) (DuPaul et al. 1998), and had a Clinical Global Impression of ADHD Severity (CGI-ADHD-S) (Guy 1976) score of ≥4 at screening and baseline.
Other inclusion criteria included use of a reliable method of birth control during the study and for 1 month following the last dose of study drug for male and female participants who were or became sexually mature; and all participants were to be of normal intelligence without evidence of significant intellectual deficit.
Exclusion criteria included weighing <16 kg at screening and baseline; history of bipolar disorder, psychosis, or pervasive developmental disorder; history of seizures or known electroencephalographic abnormalities; at serious risk of suicide in the opinion of the investigator; history of cardiovascular disease, hypertension, thyroid dysfunction, glaucoma, or urinary retention; or females who were pregnant or breast-feeding.
Study design
The study included five periods across 252 weeks of treatment followed by two follow-up safety assessments (Fig. 1). Study period (SP) I was a screening period to determine eligibility and to discontinue excluded medications. SP II was a 12-week dose titration and response assessment period. Study visits were weekly through week 4, then biweekly to week 12. Participants continued on their stable dose from the previous edivoxetine study, unless it was lower than the minimum allowed, in which case it was increased to 0.1 mg/(kg·day). The dose could be adjusted in increments or decrements of 0.1 mg/(kg·day) during the first 3 weeks of this period, and then it was to remain unchanged. At week 12, all participants were required to have a CGI-ADHD-S score of ≤3 to proceed to SP III or they were discontinued.
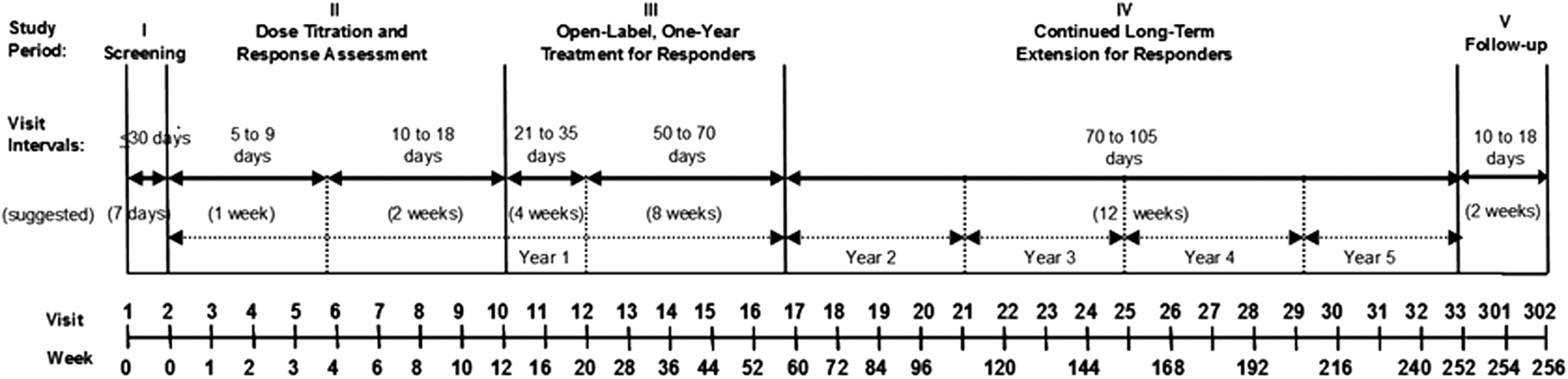
Study design.
SP III was a 1-year treatment period for responders. During this period, the dose of edivoxetine was to remain unchanged if possible, although dose adjustment was allowed at the investigator's discretion based on tolerability and efficacy as long as the maximum dose of 0.3 mg/(kg·day) and 18 mg/day was not exceeded. The first two visits in this period were 4 weeks apart, and then visits were at 8-week intervals to the end of year 1. Participants could proceed to SP IV if they tolerated edivoxetine well and the investigator felt they were benefiting from the treatment. However, the participant was discontinued from the study at any time if the investigator and/or the participant decided to discontinue.
SP IV was a long-term maintenance period for up to an additional 4 years, during which time the dose was to remain unchanged if possible, but could be adjusted up or down as described above. Visit intervals were 12 weeks apart during this SP. All participants, those who discontinued early as well as those who completed SP IV, were to enter SP V, which was a 4-week period with two required follow-up safety visits. During SP V, participants could also undergo an optional taper off edivoxetine. The drug taper schedule was 1 week on tapered dose and another week with no study drug.
Safety measures
The primary objective of this study was to assess the long-term safety of edivoxetine in children and adolescents with ADHD as indicated by serious adverse events (SAEs) reported over the duration of the study. Secondary objectives included: assessment of safety and tolerability of edivoxetine based on spontaneously reported treatment-emergent adverse events (TEAEs), discontinuation rates, discontinuations due to adverse events, and changes from baseline in electrocardiograms (ECGs), clinical laboratory measurements, vital signs, growth and sexual maturation, including height, weight, self-examined Tanner Stage in all participants (Tanner 1987), and onset of menarche in female participants. Suicide-related thoughts and behaviors were assessed using the Columbia-Suicide Severity Rating Scale (C-SSRS) (Posner et al. 2007) at every visit.
Secondary objectives also included assessment of change in ADHD symptoms using the ADHDRS-IV-Parent:Inv and the CGI-ADHD-S. ADHDRS-IV-Parent:Inv is an 18-item scale with 1 item for each of the 18 symptoms contained in the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV) diagnosis of ADHD. Each item is scored on a 0 to 3 scale (0 = never or rarely; 1 = sometimes; 2 = often; 3 = very often). The rating scale assesses the symptom severity over the past week. The CGI-ADHD-S is a single-item rating of the clinician's assessment of the severity of ADHD symptoms in relation to the clinician's total experience with ADHD patients. Severity is rated on a 7-point scale (1 = normal, not at all ill; 7 = among the most extremely ill patients).
Statistical analysis
In general, safety and efficacy analyses for the study overall were conducted on an intent-to-treat basis. Data analyzed included all enrolled participants, even if the participant did not comply with the protocol. However, for analysis within a particular study period, only participants who entered the specific study period were included. When mean change from baseline was assessed, the participant was included in the analysis only if he/she had a baseline and a postbaseline measurement.
The incidence of adverse events; discontinuations due to adverse events; and out-of-range laboratory values, vital signs, and ECG intervals were summarized. Mean changes from baseline to last observation carried forward (LOCF) endpoint for laboratory data, vital signs, and ECG intervals were also summarized with 95% confidence intervals (CIs). Mixed-model repeated measures (MMRM) analysis was also used to estimate changes from baseline in vital signs and ECG parameters at each time point during the study. Baseline and visit were independent variables in the MMRM model.
Categorical changes in blood pressure (BP) and pulse were assessed by the number and percentage of participants who had an increased BP of ≥5 mm Hg at any postbaseline time point, and that reached a value of ≥95th percentile of the United States standard for pediatric BP adjusted by age, gender, and height according to the National High Blood Pressure Education Program Working Group on Hypertension Control in Children and Adolescents (2004). Additionally, the criteria for categorical changes in pulse rate were defined as an increase of ≥25 beats per minute (bpm) from baseline and that reached to ≥110 bpm, or a pulse rate decreased by ≤15 bpm and lowered to a value of ≤60 bpm.
Treatment-emergent prehypertension BP was defined as a systolic BP of >120 mm Hg, but less than the 90th percentile BP value, or ≥90th percentile and less than the 95th percentile systolic BP values. Prehypertension diastolic BP was defined as >80 mm Hg and less than the 90th percentile value, or ≥90th percentile and less than the 95th percentile value. Treatment-emergent hypertension stage 1 systolic and diastolic BP was defined as ≥95th percentile value and ≤ to the BP value at the 99th percentile +5 mm Hg; and treatment-emergent hypertension stage 2 was defined as greater than the BP value at the 99th percentile +5 mm Hg. Sustained BP elevation was defined as an increase of ≥5 mm Hg to ≥the 95th percentile for age, gender, and height over three or more consecutive visits.
Standardized growth analyses were conducted for all enrolled participants. Percentiles of weight, height, and body mass index (BMI) were calculated based on the Centers for Disease Control and Prevention (CDC) standard growth data (released in 2000). Standardized growth data and analysis programs were provided using SAS program GC-CALCULATE-BIV.SAS obtained from the CDC website (
Analysis of improvement in ADHD symptoms were conducted utilizing LOCF and MMRM methods as described above.
Results
Participant characteristics
Participant baseline characteristics are summarized in Table 1. Participants had a mean (SD) age of 11.6 (3.0) years and were primarily male and white. The mean age at which the first ADHD symptoms were noted was ∼4.8 years with a range of 1 to 8 years, and the mean age of first ADHD diagnosis was 8.8 years, with a range of 2 to 17 years. Approximately 44% of enrolled participants exhibited signs of a learning disorder, motor skills disorder, or communication disorder.
ADHD, attention-deficit/hyperactivity disorder; ADHDRS-IV-Parent:Inv, Attention-Deficit/Hyperactivity Disorder Rating Scale-IV Parent:Investigator Rated; CGI-ADHD-S, Clinical Global Impression of ADHD Severity; n, number with characteristic; SD, standard deviation.
Participant disposition
All 267 participants who entered the study did so from another edivoxetine study and all were assigned to treatment with edivoxetine and received at least one dose of study drug. The overall mean dose (SD) was 10.4 (4.2) mg/day, ranging between 2 and 18 mg. Mean (SD) duration of exposure to edivoxetine (including exposure during lead-in study) was 528.9 (595.3) days, ranging between 3 and 2029 days.
Study completion rates across study periods are summarized in Table 2. A total of 95/267 (35.6%) participants completed 1 year of this study, and only 20/267 (7.5%) participants completed 5 years. The most common (≥10%) reasons for early study discontinuation across all study periods combined were lack of efficacy, lost to follow-up, parent/caregiver decision, adverse event, and subject decision. There were 33 participants (12.4% overall) who discontinued due to an adverse event, and fatigue (n = 4) was the most frequently reported event leading to discontinuation, followed by irritability (n = 3), vomiting (n = 3), nausea (n = 2), and tachycardia (n = 2).
N, the number of enrolled participants overall or who entered each study period.
Safety outcomes
Serious adverse events
No deaths occurred during the study. During SP II–IV, SAEs were reported by seven (2.7%) participants. Six of the seven participants reported one SAE each, specifically suicidal ideation, ankle fracture, affective disorder, appendicitis, intentional product misuse, and dengue fever. One of the seven participants reported three SAEs that included road traffic accident, contusion, and skin abrasion. During SP V, one participant was diagnosed with suspect epilepsy. None of the SAEs reported was thought to be related to study drug by the study investigators.
Treatment-emergent adverse events
A total of 203 participants (76.9%; N = 264) experienced at least one TEAE during SP II–IV (Table 3). Events reported in >10% of participants were headache, vomiting, nausea, and upper respiratory tract infection. The majority of the TEAEs were mild to moderate in severity.
N, number of evaluable participants, defined as enrolled participants who were not “lost to follow-up” who entered the study overall or each study period; n, the number of participants with at least one event per preferred term for an event overall or within the study period. Participants with repeated events per preferred across study periods would be counted only once for the overall assessment.
Discontinuation-emergent AEs
Eighty participants entered SP V to be followed up after discontinuing edivoxetine. Twelve (15.0%) participants reported discontinuation-emergent adverse events, the most frequently reported of which were upper abdominal pain (n = 2) and upper respiratory tract infection (n = 2).
Vital signs
Increases in sitting pulse of ≥25 bpm from a baseline of <110 bpm to a value of >110 bpm at any time were noted for 19.0% (47/248) of the participants. Decreases in sitting pulse of ≥15 bpm from a baseline value of ≥60 bpm to a value of <60 bpm were noted for 6.9% (18/260) of the participants.
Categorical changes in vital signs during SP II–IV are summarized in Table 4. Elevations at any time in sitting systolic and diastolic BP were reported for 35.9% (89/248) and 32.8% (81/247) of the participants, respectively. Treatment-emergent high prehypertension systolic and diastolic BP at any time was reported in 23.3% (51/219) and 19.5% (45/232) of the participants, respectively. Treatment-emergent hypertension stage 1 systolic and diastolic BP was reported in 25.6% (56/219) and 28.9% (67/232) of participants, respectively; and treatment-emergent stage 2 hypertension systolic and diastolic BP was reported in 5.5% (12/219) and 3.9% (9/232) of participants, respectively. During SP II–IV, sustained increases in sitting systolic and diastolic BP were reported in 2.5% (6/240) and 4.6% (11/240) of the participants, respectively. Mean values for heart rate and BP decreased during SP V after discontinuation of edivoxetine.
For these analyses, normal systolic BP is defined as ≤120 mm Hg and <90th percentile; normal diastolic BP is defined as ≤80 mm Hg and <90th percentile.
BP, blood pressure; mm Hg, millimeters of mercury; n, number of participants in category; N, number of participants with a normal baseline and at least one postbaseline result.
Electrocardiograms
During SP II through SP IV, there were no participants who had a QTcF increase of >60 milliseconds from baseline, and no participants who had a baseline QTcF ≤500 milliseconds and a postbaseline QTcF >500 milliseconds.
Growth
Mean percentiles for height, weight, and BMI during edivoxetine treatment are summarized by gender in Table 5. For both male and female participants, LOCF mean percentiles were similar to baseline mean percentiles for each measure.
BMI, body mass index; LOCF, last observation carried forward; SP, study period.
Sexual maturation
Tanner results were from male (N = 100) and female (N = 48) participants who had a baseline and at least one postbaseline Tanner stage score. At baseline, the participants' median age was 12.13 years, and the median exposure to edivoxetine during the study was 246 days. Based on U.S. norms for non-Hispanic white youth, age 12.13 years for females would correspond to Tanner stage 3 for pubic hair and stage 3 for breast development (Sun et al. 2002). At baseline, 54.2% (26/48) of the female participants were at pubic hair Tanner stage 3 or greater, and 42.3% (11/26) of these participants advanced one or more Tanner pubic hair stage during the study. Similarly, 54.1% (26/48) of the female participants were at breast Tanner stage 3 or greater at baseline, and 53.8% (14/26) advanced one or more Tanner breast stage.
For males, age 12.13 years would correspond to Tanner stage 2 for pubic hair and Tanner stage 3 for genital development (Sun et al. 2002). At study baseline, 54.0% (54/100) males were at pubic hair Tanner stage 2 or greater, and 64.8% (35/54) advanced one or more Tanner stage during the study. Similarly, at study baseline 51% (51.0/100) of males were at genital Tanner stage 3 or greater, and 52.9% (27/51) advanced one or more Tanner stage during the study.
Of the 75 female participants who reported their menstrual status at baseline, most (54.6%, 41/75) were premenarche. Twenty-eight of these participants reported a postbaseline menarche status, and 9 (32.1%) of these participants had experienced the onset of menses during SP II–IV.
Clinical laboratory tests
Upon review of the treatment-emergent hematology values, it was noted that 34.5% (92/267) of participants had a baseline monocyte count that was greater than or equal to the low limit at baseline. Of these, 44 (47.8%) participants had one or more treatment-emergent low values for monocyte count. It is unlikely that this observation is clinically significant because the individual participants with treatment-emergent low monocyte values did not report any unusual adverse events. There were no participants with normal baseline alanine aminotransferase (ALT) who had a treatment-emergent ALT value ≥3 × upper limit of normal (ULN), or participants with a baseline normal total bilirubin who had a treatment-emergent total bilirubin value ≥2 × ULN.
Columbia-Suicide Severity Rating Scale
Suicidal ideation assessed by the C-SSRS was reported by 15/264 (5.7%) participants during SP II–IV, but no suicidal behavior was reported. During the treatment discontinuation phase (SP V), one participant experienced suicidal ideation, but did not display suicidal behavior.
Efficacy outcomes
Overall, participants experienced improvement from baseline in ADHDRS-IV-Parent:Inv total and subscale scores, and in the CGI-ADHD-S (Table 6).
LOCF analysis, all participants with at least one postbaseline assessment.
Within group comparison, mixed model repeated measures analysis.
ADHDRS-IV, attention-deficit/hyperactivity disorder rating scale; CGI-ADHD-S, Clinical Global Impression of Attention-Deficit/Hyperactivity Disorder Severity; CI, confidence interval; LOCF, last observation carried forward; LS, least square; MMRM, mixed-model repeated measures; SD, standard deviation.
Discussion
This was a Phase 2/3 open-label flexible dosing study in children and adolescents with ADHD assessing the long-term safety and tolerability of edivoxetine for up to 5 years. All participants enrolled in this study rolled over from a prior edivoxetine pediatric study. The overall study population was representative of an average pediatric ADHD study cohort, with the majority of participants being male and having an average age at first ADHD symptoms of 4.8 years. Only 20 out of 267 enrolled participants completed 5 years of edivoxetine treatment. The study was terminated early due to slow enrollment and the very low number of 5-year completers. Lilly is not proceeding with further development of edivoxetine, as announced in Eli Lilly Press Release (2013).
The findings from this study present a safety profile for up to 5 years of edivoxetine monotherapy in children and adolescents with ADHD, although the lack of a control group limits the conclusions that can be drawn from the data. No new or unexpected tolerability or safety issues were encountered with long-term edivoxetine treatment. The percentage of participants with SAEs was low (2.7%), and the SAEs reported were not thought by investigators to be related to edivoxetine. TEAEs were broadly similar to those previously reported in randomized controlled trials of edivoxetine in children and adolescents with ADHD (Jin et al. 2013; Lin et al. 2014), in an adult population with major depressive disorder (MDD) (Pangallo et al. 2011), and observed in a long-term, open-label safety study of edivoxetine as adjunctive treatment for adults with MDD (Ball et al. 2015). Similarly, in a pooled analysis of long-term (≥3 years) atomoxetine treatment, headache, nasopharyngitis, and vomiting were the most common TEAEs reported (Donnelly et al. 2009).
Increases in BP and pulse were observed across visits during the open-label edivoxetine treatment, with mean values for heart rate and BP decreasing during SP V after discontinuation of edivoxetine. A total of 6/240 and 11/240 participants experienced sustained increases in systolic and diastolic BP, respectively. In long-term atomoxetine pooled data, a mild decrease in heart rate was observed, which was not considered to be clinically significant. Despite statistically significant pressor effects in the long-term atomoxetine studies, end-point values of an increase in systolic/diastolic BP of ∼8.4/3.2 mm Hg was deemed consistent with the increases observed in the general population from age 10 to 15 years (Donnelly et al. 2009). Similar to what we have observed in this study with edivoxetine, previous studies had demonstrated that pressor effects of atomoxetine tend to occur early in therapy, after which BPs stabilize, and then return toward baseline upon drug discontinuation (Wernicke et al. 2003).
Growth percentiles for male and female participants were overall stable throughout treatment, indicating similar rates of physical development in this pediatric cohort during treatment with edivoxetine compared with national age-matched cohorts. Assessment of the effect of edivoxetine treatment on sexual maturation is limited by the small sample size, participant drop-outs, and a relatively short median exposure time. Nevertheless, over 30% of participants had a clinically meaningful advance in sexual maturation (increased Tanner stage or onset of menses) over a median edivoxetine exposure time of 246 days. Similarly, Spencer et al. (2007) reported that continuous atomoxetine treatment for up to 5 years had little or no long-term effect on juvenile growth and final stature for most participants. An 18-month study in pediatric ADHD also showed that long-term atomoxetine treatment was not associated with any appreciable impact on or delay in sexual maturation in children with ADHD compared with U.S. normative data (Trzepacz et al. 2011).
Due to the open-label study design and low number of participants completing the study, minimal interpretation of the efficacy analyses is possible. Results were, however, included for completeness.
Conclusion
Long-term open-label treatment with edivoxetine as monotherapy in children and adolescents with ADHD revealed a safety profile that was consistent with its pharmacological effects on norepinephrine transmission and with that reported in shorter duration studies of edivoxetine.
Clinical Significance
ADHD often needs to be treated with medications for long term. The present study reports safety data from long-term treatment with edivoxetine, a selective norepinephrine reuptake inhibitor, in children and adolescents with ADHD.
Footnotes
Disclosures
All authors are full-time employees of Eli Lilly and Company. Authors E.S.M.N., M.B., P.L., and J.A. are employees of Eli Lilly and Company, Indianapolis, IN. Author D.P. is an employee of Lilly Research Center, Windlesham, Surrey, United Kingdom, a subsidiary of Eli Lilly and Company, Indianapolis, IN.