
Abstract

Chief Complaint and Presenting Problem
A.
History of Present Illness
Mother reported that A. had a history of separation anxiety as a young child, and a brief period of depressive symptoms when she was ∼14 years old, but these resolved spontaneously and did not require psychiatric treatment. A. was referred at age 16 for generalized anxiety symptoms, full-symptom panic attacks, and secondary depressive symptoms. Specifically, she endorsed irritability, restlessness, dreadful anticipation, and uncontrollable worries regarding school, her family, her brother, the future, and her friends, which occurred for several hours each day. In addition, she experienced initial insomnia, often lying in bed at night worrying about the next day, the events of the previous day, and her future. Her panic attacks occurred several times each week and included a myriad of somatic and cognitive symptoms, such as dyspnea, palpitations, nausea, tremulousness, sweating, and a sense that she might die.
A.'s symptoms had been occurring for nearly a year, but had intensified recently, prompting her and her family to seek treatment from her primary care physician. She was initially treated with sertraline 25 mg and titrated to 100 mg daily, and lorazepam 1 mg daily on an as needed basis. She was also referred to a psychotherapist. Pharmacotherapy and psychotherapy were started in parallel, given the severity of the symptoms. Despite these efforts, A.'s anxiety and panic symptoms increased, and she became unable to attend school. At that time, the only precipitant was significant distress surrounding her brother's diagnosis of bipolar disorder, although it was noted that, contemporaneously, her family attempted to accommodate her anxiety to minimize associated familial conflict. A. denied obsessive-compulsive symptoms, symptoms of mania (other than irritability), and psychotic symptoms. A. was subsequently referred to the emergency department, and treatment in a partial hospitalization program was recommended.
During the course of A.'s partial hospitalization, sertraline was titrated to 200 mg daily and lorazepam 1 mg was continued pro re nata for panic attacks. In addition, hydroxyzine 50 mg twice daily and melatonin 6 mg were added to target anxiety and dyssomnia, respectively. Continuing to struggle with severe anxiety, feeling overwhelmed with regard to falling further behind at school, and having “failed,” A. developed suicidal ideation (Columbia Suicide Severity Rating Scale [CSSRS] Intensity scores of 1–2) (Posner et al. 2011). Based on concerns that she had developed increasing depressed mood, social withdrawal, some anhedonia, and increased guilt—consistent with a diagnosis of major depressive disorder—extended release of bupropion was initiated at 150 mg qAM. At that time, a routine electrocardiogram revealed QT prolongation (QTc 483 milliseconds, normal range: 350–440 milliseconds) and sertraline was discontinued. Unfortunately, A.'s anxiety symptoms significantly worsened upon discontinuation of sertraline, and so her psychiatrist began duloxetine 20 mg qAM. Lorazepam, which had been administered on an as needed basis, was changed to scheduled clonazepam 1 mg BID. A. continued to report significant anxiety, remained unable to attend most of her classes at school, and began to feel increasingly hopeless. She developed nonsuicidal self-injurious behavior, superficially cutting on her arms. Duloxetine was increased to 40 mg/day and bupropion was discontinued, after A. had developed a low-amplitude tremor, which was attributed to the bupropion.
Following discharge from the partial hospitalization program, in the context of a number of school-based interventions and intensification of psychotherapeutic efforts, A.'s anxiety had transiently improved, and she was able to return to school part-time. However, after her return to school, there was a profound recrudescence of her anxiety and suicidal ideation (intensity score of 3 on the CSSRS) and A. was admitted to the inpatient psychiatric unit. Duloxetine was continued; clonidine was added at 0.1 mg BID and lisdexamfetamine was initiated at 30 mg daily for ADHD symptoms (e.g., inattention, distractibility, poor attention to details, and poor task persistence). However, A. was subsequently switched to methylphenidate OROS because her QT interval had again prolonged (476 milliseconds).
Following 5 days on the inpatient unit, A. was discharged on a regimen of duloxetine 40 mg qAM, clonidine 0.1 mg BID, methylphenidate OROS 36 mg qAM, clonazepam 1 mg BID PRN, and melatonin 6 mg at bedtime. Her QT interval remained borderline prolonged (440 milliseconds), but methylphenidate was continued. Over the course of several months, A.'s duloxetine was titrated to 90 mg qAM, methylphenidate OROS was increased to 72 mg qAM, and to target persistent anxiety, gabapentin was initiated at 300 mg BID. Clonidine was continued at 0.1 mg BID and no changes were made to her clonazepam regimen. She continued in psychotherapy, but reported ongoing anxiety and anxiety-related insomnia, for which zolpidem was initiated at 5 mg QHS. Over the course of a year, A. experienced worsening depression and anxiety triggered by ongoing family stressors (e.g., worsening of her brother's affective disorder and subsequent psychiatric hospitalization) and was beginning to miss school. Extended release bupropion 150 mg qAM was added to her medication to address depressive symptoms; however, due to worsening anxiety, it was discontinued after a few weeks.
In parallel, given that A. had several episodes of increasing QT prolongation with multiple psychotropics (sertraline and lisdexamfetamine), consultation with cardiology was sought. Generally, she was asymptomatic from a cardiac standpoint, aside from mild shortness of breath during sports and episodes of “chest tightness” in times of anxiety. She denied presyncope, syncope, and seizures and had never had a cardiac arrest. Over the course of her cardiac workup, electrocardiograms were performed on A.'s mother and father. Mother's QTc was prolonged at 465 milliseconds. A.'s Schwartz score was ≥3.5, consistent with classification as high risk for long QT syndrome, and thus, genetic testing was performed. A splice site mutation (c477 + 5 G>A) in the KCNQ1 gene consistent with LQT1, a genetic form of long QT syndrome, was identified. Further testing demonstrated the same mutation in three maternal aunts; two of these aunts had QTc intervals longer than 500 milliseconds. A. was subsequently started on nadolol 20 mg every 12 hours for ventricular arrhythmia prophylaxis and was titrated to 60 mg/day. She tolerated this medication well.
A. was referred to our clinic, given the persistence of anxiety symptoms despite numerous psychopharmacologic interventions and ongoing psychotherapy.
Past Psychiatric History
Beginning at age 16, A. was treated with cognitive behavioral therapy approximately every other week, for anxiety and depressive symptoms and reported a good alliance with her therapist.
Developmental History
A. was the product of a 40-week uncomplicated pregnancy and spontaneous vaginal delivery. Developmental milestones were reported to be within normal limits. Mother reported that A. had difficulty transitioning to preschool because of anxiety.
Educational History
A. attended a private high school and did very well. However, accommodations were needed for her anxiety; she received extra time for some assignments and attended school half-days.
Social History
A. lived with her mother, father, and older brother, who often experienced major mood symptoms related to bipolar disorder. A. often worried about his well-being. A. was in a long-term relationship with a boy of her same age. There was no history of abuse or neglect. A. denied alcohol or drug abuse and was not sexually active.
Family History
Family history was remarkable for major depressive disorder in mother and bipolar disorder in her brother. Two paternal second degree relatives had anxiety disorders.
Mother and three maternal aunts had long QT syndrome diagnosed following A.'s positive genetic testing. One maternal second degree and a third degree relative had histories of premature ventricular contractions, and one maternal second degree relative developed atrial fibrillation in her 20s. There was no known family history of pacemakers, implantable cardioverter defibrillators, sudden cardiac death, or cardiomyopathies.
Medical History
A. was generally a healthy adolescent, although her BMI was in the 90th percentile. She had no history of serious medical problems or hospitalizations. There was no history of thyroid disease or anemia, and she had normal renal and hepatic function. There was no evidence of impaired glucose metabolism or insulin insensitivity. Pharmacogenomic testing revealed that A. was phenotypically an extensive metabolizer for CYP2C19 (*1/*1) and was an intermediate metabolizer for CYP2D6 (*1/*4, no duplication).
Mental Status Examination
A. appeared somewhat older and more mature than her stated age. She was anxious appearing as she sat on the couch in the consultation room, and there was mild psychomotor agitation consisting of fidgeting with her hands and tapping of her legs. Moreover, despite a veneer of confidence, there was an anxious quality to her presence in the room. Speech and language were within normal limits, but remarkable for cautiousness in her choice of words, and precision in terms of her sentence structure. Her mood was “good,” although her affect was markedly anxious and defensively bright. Her thought processes were linear and goal directed. Her thought content was notable for thoughts of death and “not being alive,” but she denied suicidal intent or plan, and she remained future oriented about treatment, her family, and long-term plans. She denied auditory and visual hallucinations and there was no objective evidence of response to internal stimuli. Attention was generally fair, and memory was intact. A. was oriented to person, place, and time. Insight was fair and judgment was age appropriate.
Outpatient Course
A. described past panic attacks and ∼50 days of school missed during the school year. Panic attacks were “full symptom” and accompanied by prominent respiratory and cardiovascular symptoms. Her Panic Disorder Severity Scale (PDS) (Shear et al. 2001) score was 21 (possible range: 0–28, scores >9 suggestive of panic disorder), and she was diagnosed with panic disorder based on DSM-5 criteria. A.'s anxiety symptoms, which she described as difficult to control, included a sense of restlessness, easy fatigability, and muscle tension. She had a history of marked and persistent fear and significant anxiety in social situations related to embarrassment and humiliation. She reported that these social anxiety symptoms were now minimal, but continued to be accompanied by a tendency to avoid social situations. Her Generalized Anxiety Disorder 7 Scale (GAD-7) score was 11 (possible range: 0–21; scores of 5, 10, and 15 correspond to mild, moderate, and severe levels of GAD symptoms, respectively). A.'s ADHD symptoms remained well controlled on methylphenidate OROS.
Given that A.'s anxiety had ultimately only responded to sertraline and was minimally—if at all—responsive to α2 agonists, benzodiazepines, duloxetine, and bupropion, it was decided in consultation with cardiology, the patient, and the family to proceed with a cross-titration of duloxetine to sertraline. Sertraline was initiated at 25 mg daily, and duloxetine was continued at 90 mg daily. A. continued on gabapentin 600 mg twice daily and zolpidem 5 mg nightly as needed for initial anxiety-related insomnia. At the time, she was not attending school and was having several panic attacks weekly. Clonazepam was initiated at 0.25 mg twice daily during the duloxetine-sertraline cross-titration.
Over the course of 2 months, sertraline was titrated in 25 mg increments to 150 mg daily. Serial electrocardiograms were obtained and the QTc interval varied with sertraline dosage (Fig. 1). A. first reported decreasing anxiety and increasing school attendance when sertraline was titrated to 100 mg daily. At that time, she had had only one panic attack over the preceding 2-week period, and her PDS score had decreased from 21 to 8. When sertraline was increased to 100 mg daily, duloxetine was gradually discontinued over 3 months in 30 mg decrements. Concomitantly, A.'s PDS score decreased to 3 and her GAD-7 score improved from 11 to 5 (Fig. 1).
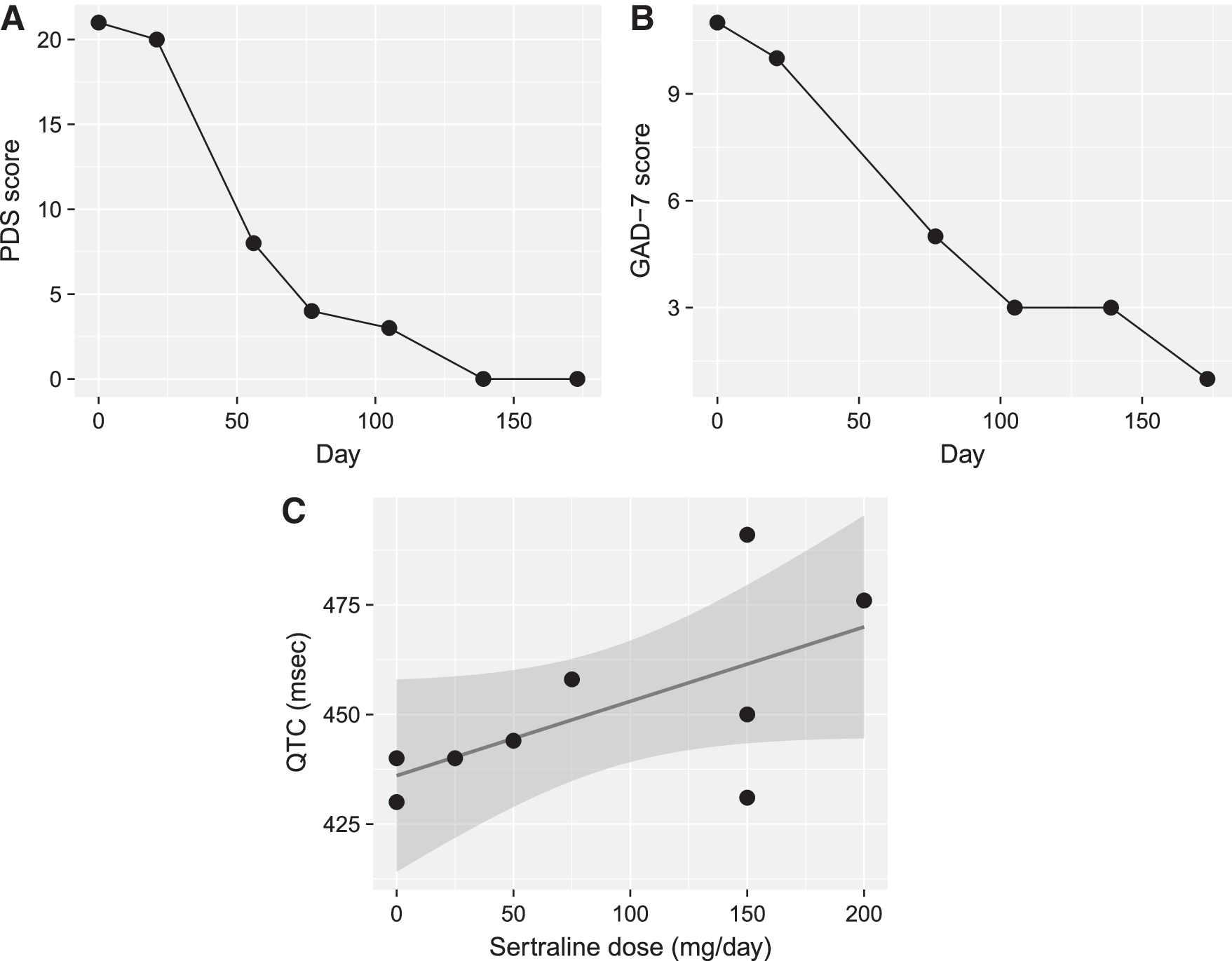
Patient outcomes during sertraline treatment.
At this time, as her anxiety had improved significantly, A. also reported the evolution of sedation, amotivation, and fatigue, which were thought to be secondary to gabapentin or clonazepam. Gabapentin was decreased and discontinued, which led to improvement in energy and motivation. Clonazepam was reduced to an as needed basis (0.5 mg twice daily) and zolpidem was discontinued. After 4 months on sertraline monotherapy, A.'s PDS score remained 0 and her GAD-7 score 1 (Fig. 1). She continued to do very well from an anxiety standpoint. Her ECG demonstrated acceptable corrected QT intervals with no concerning cardiac symptoms to date.
Brief Formulation
A.'s anxiety and depressive symptoms had been minimally responsive to both psychotherapeutic and psychopharmacologic interventions. Moreover, she experienced a significant symptom burden, panic attacks almost daily, and struggled interpersonally with anxiety. She met DSM-5 criteria for generalized anxiety disorder, panic disorder, social anxiety disorder, and attention-deficit/hyperactivity disorder. Notably, her most robust treatment response for anxiety was with sertraline; however, this was discontinued secondary to her prolonged QT interval.
From a biological perspective, A. had a genetic vulnerability for both affective and anxiety disorders, and for QT prolongation. From a psychological standpoint, there was an early history of anxious–ambivalent attachment and behavioral inhibition, which increased her risk of developing an anxiety disorder (Delgado et al. 2015). From a psychosocial standpoint, A. was initially referred during a time of significant distress surrounding her brother's diagnosis of bipolar disorder, and in parallel, her family had attempted to accommodate her anxiety to minimize associated familial conflict.
Multiaxial Diagnoses
Axis I: Generalized anxiety disorder
Panic disorder with agoraphobia
Social anxiety disorder
Attention-deficit/hyperactivity disorder, inattentive type
Major depressive disorder, past
Axis II: Deferred
Axis III: Congenital long QT syndrome.
Obesity
Axis IV: Problems with primary support; educational problems
Axis V: Current Global Assessment of Functioning score: 50 at the beginning of treatment
Discussion
This case illustrates the complexities and challenges of treating anxiety in an adolescent with susceptibility to QT interval prolongation. Anxiety disorders are among the most common psychiatric conditions in children and adolescents, increase the risk of secondary disorders, and are associated with significant morbidity (Beesdo et al. 2009). Moreover, selective serotonin reuptake inhibitors (SSRIs) are the primary psychopharmacologic treatments for youth with anxiety disorders (Connolly and Bernstein 2007), but may be associated with a number of adverse events (Reinblatt et al. 2009; Strawn et al. 2015). Among these adverse events associated with several antidepressants is the risk of QT prolongation, particularly in vulnerable individuals.
Congenital long QT syndrome—an autosomal dominant disease resulting from mutations in gene encoding cardiac ion channels—has a prevalence of ∼1 in 2500, and over 600 mutations have been identified in 16 long QT syndrome (LQTS) genes (Schwartz et al. 2012; Barsheshet et al. 2014). Mutations in these genes tend to prolong the duration of the ventricular action potential, thus lengthening the QT interval, predisposing to early after-depolarization, and ventricular arrhythmias. Ventricular arrhythmias can lead to syncope, seizures, and sudden cardiac death in children and adults without structural cardiac disease. In addition, mutations in the KCNQ1 gene, causing the LQT1 phenotype, are present in ∼40%–55% of patients with autosomal dominant LQTS and accentuate the risk of arrhythmia secondary to sympathetic activation (Schwartz et al. 2012; Barsheshet et al. 2014). Overall, ∼25% of patients with genetically confirmed LQTS have QTc intervals well within the normal range (Goldenberg et al. 2011). Of individuals with the LQT1 phenotype, 12% have QTc ≤440 milliseconds. In addition, the genetic disorder exhibits incomplete penetrance (not all patients have phenotypic expression) and variable expressivity (phenotypic expression differs among patients). Up to 37% with the LQT1 phenotype remain asymptomatic. Patients with a history of prior syncope or QTc greater than 500 milliseconds are considered high risk, whereas patients with QTc duration ≤500 milliseconds without prior syncopal event are considered lower risk (Barsheshet et al. 2014).
QT prolongation in association with psychiatric medications has received increased attention over the last decade; patients with familial LQTS would be at particular risk on SSRIs that may be associated with QT prolongation (Table 1).
Substantial evidence supports the conclusion that these drugs prolong the QT interval and are clearly associated with the risk of TdP, even when taken as directed in official labeling.
Substantial evidence supports the conclusion that these drugs can cause QT prolongation, but there is insufficient evidence at this time that these drugs, when used as directed in official labeling, are associated with a risk of causing TdP.
Substantial evidence supports the conclusion that these drugs are associated with a risk of TdP, but only under certain conditions (e.g., “excessive” dose, hypokalemia, congenital long QT, or by causing a drug–drug interaction that results in excessive QT interval prolongation).
A. had struggled with significant anxiety despite substantial efforts in psychotherapy, and while she had initially done very well with one particular SSRI (sertraline), this SSRI was associated with a risk of QT prolongation and torsades de pointes under certain conditions (excessive dose, hypokalemia, and congenital long QT) (Woosley and Romero 2015).
While there are no prospective large studies evaluating QT prolongation in youth treated with SSRIs, several studies have examined this issue in adults, and one study has examined cardiac events in youth treated with SSRIs. In this study, pediatric patients treated with SSRIs (N = 113,714) were evaluated from health insurance claim data with regard to subsequent incidence of ventricular arrhythmias, cardiac arrest, and sudden death over a 12-month period. While QT prolongation was not directly addressed in this study, the incidence of these cardiac events was relatively low (9.8 events per 10,000 person-years) and was lowest for patients treated with fluoxetine. The incidence rate for sertraline was 10.3 per 10,000 person-years (95% CI: 6.1–17.4), with the mean time to cardiac event being 97 days. The adjusted risk of adverse event relative to fluoxetine (lowest incidence) was low for sertraline (HR = 2.14, 95% CI: 0.75–6.16) (Czaja et al. 2013). In adult patients (N = 1692) treated with SSRIs, a recent meta-analysis revealed a modest, but statistically significant increase in the QTc interval (+6.10 milliseconds, 95% CI: 3.47–8.73), although to a lesser extent than tricyclic antidepressants (Beach et al. 2014).
While citalopram is perhaps the most well-known agent with risk of QTc prolongation (Celexa® [package insert] 2014), the risk of QTc prolongation with sertraline suggests a more modest risk (Beach et al. 2014). In this regard, data from adults treated with sertraline generally suggest that clinically significant QT prolongation occurs primarily with concomitant administration of other QT-prolonging medications; thus, sertraline is thought to have a low risk for QT prolongation (Funk and Bostwick 2013). However, substantial evidence supports the conclusion that sertraline is associated with a risk of torsades de pointes, but only under certain conditions (e.g., “excessive” dose, hypokalemia, congenital long QT, or by causing a drug–drug interaction that results in excessive QT interval prolongation) (Woosley and Romero 2015).
Lifestyle modifications and β-blockers are recommended for all patients with a diagnosis of LQT1. Titration to the maximum dose appropriate for age and weight, if tolerated, is recommended for β-blockers. Patients with LQT1 have most of their events during exercise and, therefore, should avoid strenuous exercise, particularly swimming, and those at intermediate or high risk should not engage in competitive sports (Maron and Zipes 2005; Barsheshet et al. 2014).
To the authors' knowledge, no other case reports have addressed the use of SSRIs in adolescent patients with congenital long QT syndrome. In A.'s case, discussions of the risks and benefits of sertraline were extensive and involved the patient, her parents, her psychiatrist, her psychotherapist, and her cardiologist. Ultimately, control of A.'s anxiety and panic disorder was best achieved with sertraline, and her QTc remained generally ≤460 milliseconds on a therapeutic dose of sertraline.
Footnotes
Acknowledgment
The authors acknowledge and thank Maxwell Luber for his assistance in review and preparation of the article.
Disclosures
Dr. T.B.M. reports no biomedical conflicts of interest. Dr. J.R.S. has received research support from the National Institute of Mental Health, Eli Lilly, Lundbeck, Shire, Forest Research Institute, Edgemont Pharmaceuticals, and Neuronetics. He receives royalties from Springer Publishing for two textbooks. Dr. D.S.S. has received research support from Medtronic. Dr. B.J.C. has received research support from Astra Zeneca, Auspex/Teva, Catalyst, Neurocrine, NIMH, Otsuka, Shire, and Genco Sciences. Dr. J.R. and Dr. B.G.-D. report no biomedical conflicts of interest.