
Abstract
Amphetamine (AMP), an indirectly acting psychostimulant approved for the treatment of attention-deficit/hyperactivity disorder (ADHD) in children, adolescents, and adults, is among the most long-standing therapeutic agents in all of clinical psychopharmacology. This review focuses on AMP absorption, metabolism, and elimination brought to bear on comparative pharmacokinetics in its various formulations. A comprehensive search of the published literature was conducted using MEDLINE (PubMed) and Google Scholar databases through April 2017 to retrieve all pertinent in vitro and human studies for review and synthesis. Additionally, Food and Drug Administration (FDA) databases were accessed for otherwise unavailable data when possible. Initially available as racemic (dl)-AMP, this drug was later supplanted by enantiopure (d)-AMPH or enantioenriched (75:25 dl)-AMP formulations; although racemic AMP returned as an approved drug to treat ADHD in 2014. Presently, there are several immediate-release (IR) formulations available, including d-AMP, dl-AMP, and mixed amphetamine salts, which are neither racemic nor the pure d-enantiomer (i.e., a 3:1 mixture of d-AMP and l-AMP). Furthermore, new modified-release AMP formulations, including an oral suspension and an orally disintegrating tablet, are now available. A lysine-bonded prodrug form of d-AMP also serves as a treatment option. Oral AMP is rapidly absorbed, with high absolute bioavailability, followed by extensive metabolism involving multiple enzymes. Some metabolic pathways exhibit stereoselective biotransformations favoring the l-isomer substrate. Drug exposure exhibits dose-proportional pharmacokinetics. Body weight is a fundamental determinant of differences in observed AMP plasma concentrations. IR formulations typically provide a T max from 2 to 3 hours. In replicated studies, children exhibit a shorter plasma T 1/2 (∼7 hours) relative to adults (∼10 to 12 hours). There are few documented pharmacokinetic drug interactions of clinical significance beyond influences of drug-induced alteration of urinary pH. The array of AMP formulations addressed in this review offer flexibility in dosing, drug onset, and offset to assist in individualized pharmacotherapy of ADHD.
Introduction
Attention-deficit/hyperactivity disorder and the advent of psychostimulants as first-line pharmacotherapy
W
Amphetamine is a contraction of the chemical name alpha-methylphenethylamine. Approximately 80 years ago, Bradley (1937) first reported the clinical benefits of dl-AMP sulfate in child psychiatry.
Early investigations pioneered correlations, now termed pharmacokinetic-pharmacodynamic relationships, to encompass observed interindividual differences in response relative to individualizing dosing in achieving optimal benefits in behavioral response (Peoples and Guttman 1936; Sargant and Blackburn 1936). Peak effects were found to occur at 1.5 hours relative to 2.5 hours postadministration (Sargant and Blackburn 1936). In due course, these clinical observations on maximal pharmacodynamic effects were correlated with the time to maximum concentrations (T max) of AMP upon development of truly quantitative analytical methodologies. Thus, rudimentary pharmacokinetic studies of AMP provided fundamental insights into the disposition of AMP and its two isomers (Beyer and Skinner 1940).
The present review focuses on the pharmacokinetics of current AMP formulations, a drug which has been in continuous clinical use longer than any other pharmacological agent employed in modern psychiatry (Markowitz and Yu 2016). Additionally, it is one of the most extensively studied therapeutic compounds in the biomedical literature. Indeed, a PubMed search of the term “amphetamine” conducted in March of 2017, which excluded the terms “abuse” and “dependence,” yielded >31,600 citations. Accordingly, the emphasis here is more narrowly placed on the range of formulations and their relative AMP isomer contents in the context of distinguishing pharmacology of one AMP product from another.
Search methods
In preparation of this review, a computerized systematic literature search was conducted in MEDLINE (PubMed) and Google Scholar databases through April 2017 to retrieve all pertinent studies and reviews. Additional cross-referencing of published bibliographies expanded our search. These were limited to human data and in vitro data largely limited to drug metabolism studies. Animal studies were generally excluded. Indexing terms utilized included amphetamine, dextroamphetamine, d-amphetamine, levoamphetamine, l-amphetamine, LDX, pharmacokinetics, metabolism, and absorption. Only articles published in the English language were evaluated. Although it was our preference to only cite and include data published in peer-reviewed periodicals, in the interest of a comprehensive coverage of all available AMP formulations, it was necessary to include data from product labeling materials as well as online documents available through the U.S. Food and Drug Administration (FDA), Center for Drug Evaluation and Research (CDER), filed in compliance with the drug approval process. In the case of the racemic AMP reemergence after a half-century hiatus, the limited pharmacokinetic data were provided to us following a personal request (Arbor Pharmaceuticals, LLC; July 1, 2016 personal communication).
AMP isomers
The preponderance of mechanistic evidence underlying the psychostimulant effects of AMP supports an AMP-induced increase in extracellular dopamine and norepinephrine mediated by efflux of the cytoplasmic monoamines through their respective transporters. Comparisons of d- versus l-AMP isomer potency employing a variety of approaches reveal that d-AMP is anywhere from three- to seven-fold more potent than l-AMP at the dopamine synapse (Coyle and Snyder 1969; Thornburg and Moore 1973; Heikkila et al. 1975; Richelson and Pfenning 1984; Kula and Baldessarini 1991; Easton et al. 2007) and has been reported to be from one to two times as potent at the norepinephrine synapse (Coyle and Snyder 1969; Thornburg and Moore 1973; Richelson and Pfenning 1984). Whether these bench findings translate to directly corresponding clinical differences in isomer pharmacology is unclear. While AMP serves as the prototypic inducer of locomotor activity in model models, quizzically, the separate isomers have been shown to elicit opposing action spontaneous locomotor effects (Zabik et al. 1978).
Initial studies suggested that subpopulations of children with ADHD have a better clinical response when receiving racemic AMP, while others responded better to the pure isomer, d-AMP (Bradley 1950). Subsequently, clinical studies assessed the psychopharmacology of the separate in d- and l-AMP isomers, finding greater clinical benefits in controlling ADHD for each isomer, depending on the individual patient (Arnold et al. 1972, 1976). Additionally, subtle differences in adverse effects between the two isomers were reported (Arnold 2000). These earlier observations of differential effects for each AMP isomer on separate populations of ADHD children were subsequently replicated (Mcintyre et al. 1981). In any case, the majority of available AMP formulations contain both isomers.
AMP formulations
AMP is presently marketed in a number of different salts and ratios of stereoisomers delivered by a variety of oral dosage formulations. These include immediate-release (IR) tablets, extended-release tablets and capsules, an orally disintegrating tablet (ODT), and at least one prodrug formulation (Tables 1 and 2).
b.i.d., two times per day; CDER, Center for Drug Evaluation and Research; ER, extended-release; IR, immediate-release; LDX, lisdexamfetamine; ODT, orally disintegrating tablet; PI, package insert; q.d., once a day.
ADHD, attention-deficit/hyperactivity disorder; BED, binge eating disorder; ODT, orally disintegrating tablet.
The enantiopure d-AMP sulfate formulation (Dexedrine®) was FDA approved as an IR tablet, followed by a modified-release (MR) capsule (i.e., Spansule®) approved in 1976. There is little published data on the pharmacokinetics of the Dexedrine Spansule. However, it was used as a comparator formulation in pharmacokinetic studies submitted to the FDA during the course of the approval process of the d-AMP prodrug LDX. Accordingly, its fundamental pharmacokinetic parameters can be found within the basis of approval by the CDER documents (U.S. Food and Drug Administration and Center for Drug Evaluation and Research [CDER] 2006). The pharmacokinetic parameters are presented in Table 1. This formulation contains IR and MR AMP beads to permit once-daily dosing.
For the period between the early 1980s and mid-1990s, d-AMP formulations were the primary AMP entities available. In 1994, mixed amphetamine salts (MAS) were marketed as Adderall® (formerly prescribed as the obesity treatment Obetrol®). This mixture contained racemic dl-AMP sulfate, four diastereomeric AMP RS-aspartates (SS/RS/RR/SR), and two enantiopure d-AMP salts (sulfate and D-saccharate). The 20 mg strength of MAS contains 9.5 mg of d-AMP and 2.3 mg of l-AMP calculated as the free base (76%:24%). Thus, as generally perceived and described in the literature as a mixture of four AMP salts (Popper 1994). In reality, MAS exists as eight different AMP salts: the 2 enantiomers composing racemic dl-isomer AMP sulfate, the 4 diastereomers composing dl-AMP RS-aspartate (i.e., SS/RS/RR/SR), and the 2 enantiopure d-AMP salts (sulfate and D-saccharate). The reasoning behind the inclusion of disparate amounts of d- and l-AMP, and the range of salt forms incorporated, appears to have been one more of pharmaceutical convenience than scientific rationale. However, these various salts will exhibit a range of aqueous gastric dissolution rates, where even a pure isomer such as d-AMP sulfate can be expected to dissolve more rapidly than racemic dl-AMP sulfate, the so-called double solubility rule (Patrick and Straughn 2016). Extending the time course for complete MAS dissolution in the stomach through administration of these multiple AMP salt forms potentially offers a therapeutic advantage over d-AMP sulfate by consequently prolonging the AMP absorption phase. This specific portion of an AMP pharmacokinetic profile has correlated with maximal AMP efficacy in the treatment of ADHD (Brown et al. 1980).
For the first time since 1971, in September of 2014, racemic AMP sulfate reentered the clinical arena marketed under the proprietary name of Evekeo™ with an indication for the treatment of ADHD in children >3 years of age and now an AMP FDA approved for short-term exogenous obesity (Arbor Pharmaceuticals, LLC; July 1, 2016 personal communication).
Among the newer AMP formulations are products intended for patients who have difficulties swallowing standard capsules or tablets, including an MR-ODT containing MAS (AMP XR-ODT), again containing an approximate 3:1 ratio of d- and l-AMP and providing a similar pharmacokinetic profile to MR-MAS (Stark et al. 2016), as well as an MR oral suspension containing approximately a 3:1 ratio of d- and l-AMP, as well as an IR solution containing d-AMP sulfate. These differences in formulation technologies result in a range of characteristic pharmacokinetic profiles, offering drug exposure parameters distinguishing one product from another. The majority of AMP pharmacokinetic studies either published or available through the FDA have been conducted in healthy adult subjects without ADHD rather than children or adolescents diagnosed with this disorder. The specific pharmacokinetic parameters derived from many of these studies associated with different formulations are provided in Table 1 for comparative purposes. Unless otherwise noted, the following detailed section will generally refer to the oral administration of IR racemic AMP, MAS, or d-AMP. Although pharmacodynamic differences in AMP isomers have been consistently demonstrated, there are few significant differences in the primary pharmacokinetic parameters of AMP isomers that have been noted when administered in the same dosage and formulation, although enantioselective metabolic pathways have been reported (Patrick and Markowitz 1997).
AMP absorption and distribution
After oral administration of AMP, most of the dose (>90%) is absorbed. AMP does not appear to be a substrate or inhibitor of the enteric efflux P-glycoprotein transporter (Zhu et al. 2006, 2008). Product labeling information for many AMP formulations reports that gastrointestinal acidifying agents (e.g., guanethidine, reserpine, ascorbic acid, and some fruit juices) may lower absorption of AMP, while gastrointestinal alkalinizing agents (e.g., sodium bicarbonate) may increase absorption. However, the influence of the coadministration of AMP with agents, which either decrease or increase gastrointestinal pH such as antacids and H2-receptor antagonists (e.g., famotidine), has not been thoroughly investigated. A study has assessed the influence of the proton pump inhibitor omeprazole, expected to increase gastrointestinal pH, on the pharmacokinetics of the d-AMP prodrug LDX and MR-MAS. In this study, omeprazole had no significant influence on the extent of absorption of either formulation (Haffey et al. 2009). It should be noted that omeprazole and its metabolites are recognized inhibitors of both CYP2C19 and CYP3A4 (Shirasaka et al. 2017), but neither of these enzymes assumes a significant role in LDX or AMP metabolism.
The rate and extent of absorption are similar for each AMP isomer (Han et al. 1978). Food has been reported to have little effect on the bioavailability of d-AMP in either an IR formulation (Angrist et al. 1987) or earlier MR formulation, that is, Dexedrine Spansule (Brown et al. 1980). However, the consumption of food or a high-fat meal may significantly prolong the T max of both isomers in some MR dosage forms (Tulloch et al. 2002). The consumption of food concomitantly with newer generation MR-AMP formulations and prodrugs generally has no significant influence on the extent of absorption of either AMP isomer, while administration with or directly after a meal resulted in a 1–2-hour delay in attaining T max (Krishnan and Zhang 2008; Stark et al. 2016). With regard to the prodrug LDX as well as AMP XR-ODT, administration with food can decrease the C max and delay the T max of LDX and its liberated AMP, as well as from AMP XR-ODT, respectively (Adzenys XR-ODT 2016; Comiran et al. 2016). Of the MR solid dosage forms, MR-MAS capsules may be opened and sprinkled onto a food such as applesauce or yogurt, and LDX capsules can be opened and mixed with water and consumed. While the Dexedrine Spansule can be readily opened up, there is no guidance available beyond swallowing the dosage form whole.
Typically, IR T max values are reported to range between 2 and 4 hours whether administered as a racemic AMP, pure d-AMP, or MAS, although substantial intersubject variability has been reported (Han et al. 1978; Brown et al. 1979; Kupietz et al. 1985; Angrist et al. 1987; Greenhill et al. 2003; Arbor Pharmaceuticals, LLC; July 1, 2016 personal communication). While there appears to be substantial evidence of intersubject variability in AMP pharmacokinetics, data from a limited number of patients suggest that intrasubject variability is much lower (Brown et al. 1979). A general observation regarding IR AMP is that pharmacokinetic variability may be greatest during the absorption period, as opposed to the elimination phase (Brown et al. 1978, 1979). For the MR formulations of MAS (MR-MAS), a T max of 5–8 hours is typical (Tulloch et al. 2002; Clausen et al. 2005).
The buccal absorption of racemic AMP in solution has been investigated and appears to be pH dependent through passive diffusion, and absorption of d- and l-AMP was equal (Beckett et al. 1968). This finding may have relevance to the ODT d-AMP formulation.
In general, pharmacokinetic studies conducted in normal adult volunteers indicate that the area under the plasma concentration–time curve (AUC) in the 24 hours postdosing (AUC0–24) and the C max are dose proportional (Perez-Reyes et al. 1991). Following the administration of racemic AMP or the individual enantiomers, there appears to be little difference in measured pharmacokinetic parameters associated with absorption (Wan et al. 1978; Pizarro et al. 1999; Greenhill et al. 2003), although some differences in T 1/2 have been variously reported (Greenhill et al. 2003).
Therapeutic drug monitoring of AMP blood levels has not proven useful in the management of ADHD, and there appears to be little correlation between blood concentrations of AMP and behavioral effects in adults (Kupietz et al. 1985; Angrist et al. 1987) or children (Brown et al. 1979, 1980). Nevertheless, a number of studies have been conducted for the purpose of meeting regulatory requirements and in the exploration of potential pharmacokinetic–pharmacodynamic relationships. At least one small (n = 9) placebo-controlled study conducted in children treated with the MR Spansule formulation of d-AMP reported that significant clinical responses appeared to occur only during the absorption phase and were not correlated with specific plasma concentrations of the drug (Brown et al. 1980). Following doses of 0.25 or 0.5 mg/kg of IR d-AMP in healthy volunteers, C max values were ∼40 and 70 ng/mL, respectively (Angrist et al. 1987). The mean C max of d- and l-AMP following a 10-mg dose of IR-dl-AMP sulfate in healthy volunteers (n = 20) was 14.7 and 12.0 ng/mL, respectively (Arbor Pharmaceuticals, LLC; July 1, 2016 personal communication). Regarding plasma T 1/2, available data consistently show that children exhibit a significantly shorter half-life (∼7 hours) relative to those observed in healthy adults (∼10 to 12 hours) (Brown et al. 1978, 1979; Angrist et al. 1987; Tulloch et al. 2002; Greenhill et al. 2003; Arbor Pharmaceuticals, LLC; July 1, 2016 personal communication).
Addressing the d-AMP prodrug LDX, following oral administration, the pharmacologically inactive parent molecule, consisting of d-AMP covalently bonded to
The intended pharmacokinetic profile of LDX was not altered by the coadministration of the proton pump inhibitor omeprazole, which would be expected to decrease the acidity of the gastric environment (Haffey et al. 2009). It is noted that one impetus for the development of LDX was to block intranasal and intravenous abuse potential owing to the erroneous assumption that the promoiety lysine was necessarily liberated only in the gut by the hydrolytic actions of trypsin, predicted due to the knowledge that this protease cleaves lysine residues during proteolysis (Najib 2009).
The most recently FDA-approved, once-daily, MR-MAS oral dosage form appears to provide clinical coverage up to 16 hours, which is longer than any other currently approved MAS formulation. This extended duration of symptom control may be desirable in certain patients (Mydayis® 2017). This additional period of coverage is due to the prolonged drug release profile achieved through a formulation comprising three types of drug-releasing beads. This triple-bead formulation comprises an IR bead and two different types of delayed-release beads that incorporate enteric and extended-release coatings. The first delayed-release bead releases AMP at approximately pH 5.5, while the second and third delayed-release beads release AMP at approximately pH 7.0 (Ermer et al. 2007). The IR bead and the first delayed-release bead are together very similar to existing MR-MAS formulations (e.g., Adderall XR), while the third bead in the triple-bead formulation releases an additional delayed-release dose of AMP, providing extended therapeutic coverage. Thus, the formulation provides for continued release and absorption as it traverses the gastrointestinal tract.
AMP distribution
Following AMP absorption, some is believed to be taken up to some extent by red blood cells. The drug rapidly distributes into the extravascular space. AMP isomers are estimated to be ∼16% to 20% bound to plasma proteins (Franksson and Änggård 1970; Baggot et al. 1972). This relatively low protein binding is consistent with most of the drug in the bloodstream subsequently accumulating in highly perfused organs. Accordingly, AMP is widely distributed throughout the body and readily penetrates the central nervous system. Postmortem AMP cerebral spinal fluid concentrations have been reported to be ∼80% of blood (Tominaga et al. 2015).
Although both AMP enantiomers accumulate in the brain, rodent studies indicate that d-AMP may attain higher concentrations (Goldstein and Anagnoste 1965). With regard to the d-AMP prodrug LDX, rodent studies indicate that d-AMP easily crosses the blood–brain barrier, but the parent prodrug LDX does not (Hutson et al. 2014). The volume of distribution (Vd) has been estimated at 3–4 L/kg and is observed to increase with increasing body weight. Healthy adult volunteer studies have generally suggested that body weight is the primary determinant of apparent differences in the pharmacokinetics of d- and l-AMP across multiple age ranges. In general, studies have revealed dose-proportional pharmacokinetics (as indicated by C max and AUC) with most AMP dosage forms in healthy adults, adults with ADHD, and children and adolescents with ADHD (McGough et al. 2003; Clausen et al. 2005; Krishnan and Stark 2008; Boellner et al. 2010; Ermer et al. 2010).
When LDX (30–70 mg) was given as a single dose to children aged 6–12 years with ADHD, d-AMP (AUC and C max) displayed dose proportionality across the range, while concentrations of the intact prodrug did not (Ermer at al. 2010). It is noted that as a prodrug, the 70-mg dose equates to 20.8 mg of d-AMP free base.
In a study assessing the administration of each enantiomer administered separately, or as a racemic mixture under acidic or alkaline conditions, the Vd and protein binding of the isomers were similar, whereas the elimination T 1/2 of d-AMP was shorter than that of the l-isomer (Matin et al. 1977; Wan et al. 1978; Tulloch et al. 2002). Both d- and l-AMP concentrations in oral fluid (saliva) were noted to be nearly an order of magnitude higher than those measured in plasma (Matin et al. 1977), but may be proportional to plasma concentrations (Wan et al. 1978; Arbor Pharmaceuticals, LLC; July 1, 2016 personal communication). Comiran et al. (2017) recently reported that 2 hours following a 70-mg dose of LDX in adult volunteers, LDX and AMP mean concentrations were 16.3 and 136.1 ng/mL for oral fluid, respectively, and 30.6 and 39.0 ng/mL for plasma.
AMP metabolism
Much of the existing work involving the metabolism and excretion of AMP was established in the 1960s and 1970s and sought to ascertain basic metabolic pathways and contributing factors that might influence metabolism and excretion in the interest of mass balance and the determination of any potential contribution of AMP metabolites to the overall response to the orally administered drug. In the ensuing sections, the fundamental absorption, distribution, metabolism, and excretion characteristics and parameters of AMP are discussed in detail.
In man, AMP is highly metabolized and subject to two primary oxidative pathways (Fig. 1). Aromatic hydroxylation occurs at the para position to form the pharmacologically active metabolite 4-hydroxyamphetamine (i.e., para-hydroxyamphetamine) through a reaction catalyzed by CYP2D6 (Bach et al. 1999). Oxidative deamination through N- or α-hydroxylation is the other primary oxidative route and leads to the formation of phenylacetone (Dring et al. 1966, 1970; Green et al. 1986). Since CYP2D6 is highly polymorphic (Teh and Bertilsson 2012), genetic variability in AMP metabolism may be clinically relevant. However, CYP2D6 is one of several enzymes involved in the biotransformation of AMP, which potentially mitigates the overall impact of CYP2D6 genetic variants on AMP disposition. Next, para-hydroxyamphetamine is converted into p-hydroxynorephedrine through dopamine-β-hydroxylase. Additionally, β-hydroxylation occurs and is stereoselective for d-AMP to form norephedrine (phenylpropanolamine). Both 4-hydroxyamphetamine and norephedrine are pharmacologically active and subject to further metabolic oxidation. AMP deamination is believed to be catalyzed by CYP450 isoenzymes of the CYP2C subfamily (Shiiyama et al. 1997) and may proceed in a stereoselective manner. Aromatic oxidative metabolites are generally subject to secondary conjugation reactions with sulfate or glucuronic acid. Flavin-containing monooxygenase form 3 (FMO3) appears to contribute to N-oxygenation of d- and l-AMP (Cashman et al. 1999). AMP is also oxidized to phenylacetone (1-phenyl-2-propanone), which is subsequently oxidized to benzoic acid, and excreted as its glycine conjugate, hippuric acid (Dring et al. 1966, 1970).
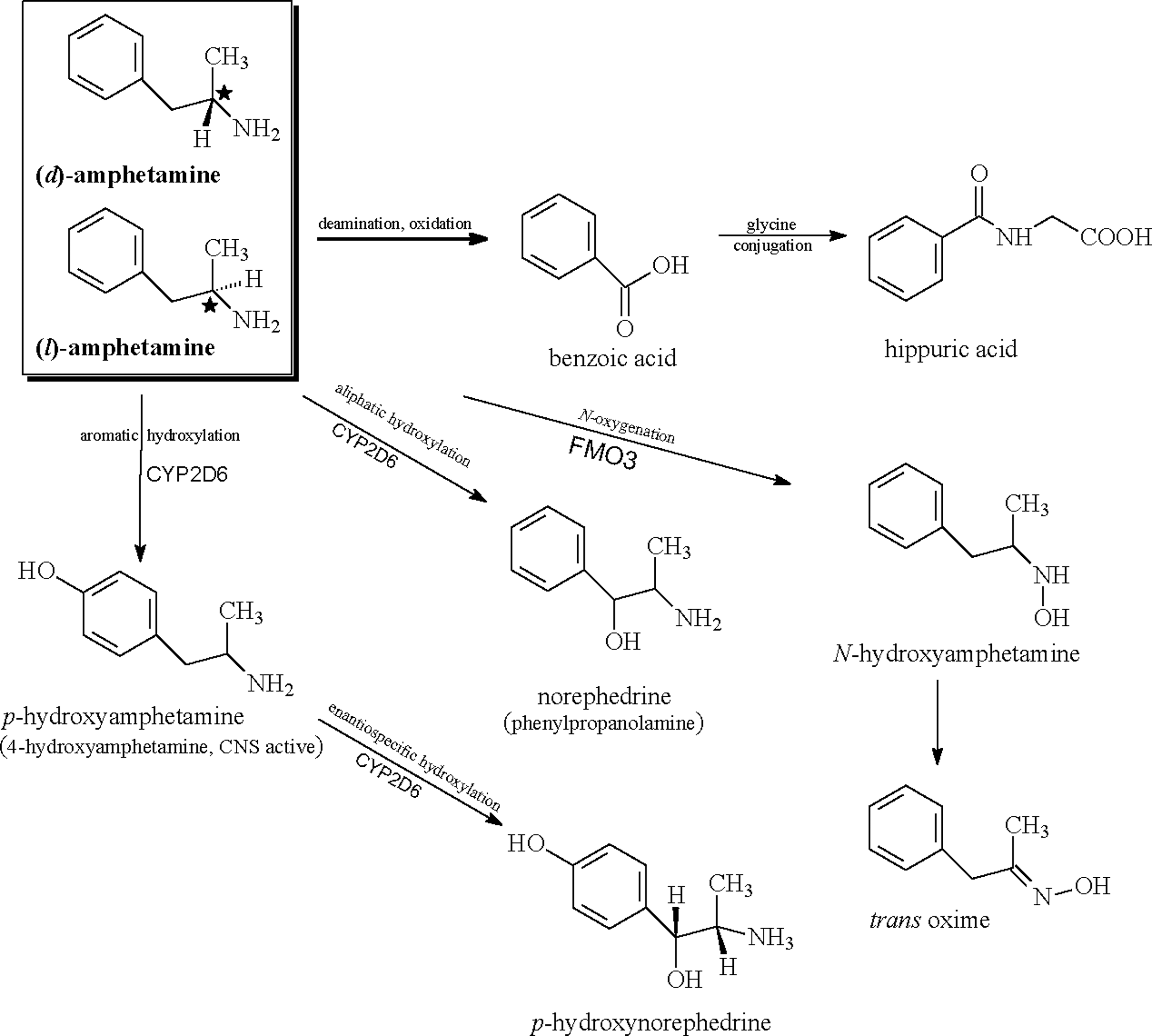
Metabolic pathways of racemic (dl)-amphetamine in man.
Stereoselective metabolism of racemic AMP was evidenced by a number of early research studies with somewhat inconsistent results and hampered by limited analytical capabilities (Cody et al. 2003). However, overall, some general observations can be made. It would appear that d-AMP is metabolized more rapidly than l-AMP, leading to differential exposure to each isomer when administered as the racemate and disproportionate concentrations of isomers excreted in the urine. Data from pharmacokinetic studies measuring plasma concentrations appear to support the findings of the preferential metabolism of d-AMP. The major difference in the disposition of the AMP enantiomers is reflected in the elimination of T 1/2 with d-AMP having a T 1/2 1–2 hours shorter than l-AMP (Wan et al. 1978; Arbor Pharmaceuticals, LLC; July 1, 2016 personal communication).
Metabolic drug–drug interactions
In vitro experiments using human liver microsomes indicate only minor to modest competitive inhibition of CYP2D6 (Wu et al. 1997; Hutson et al. 2014) and minor inhibition of CYP2A6 (Rahnasto et al. 2003) by AMP and no significant influences upon CYP1A2, 2A6, 2B6, 2C9, 2C19, 2D6, and 3A4/5 by intact LDX (Krishnan and Moncrief 2007). At least one clinical study has been carried out in normal volunteers utilizing the four-probe drug, Cooperstown cocktail, and found no evidence of significant inhibition of CYP1A2, CYP2D6, or CYP3A by LDX. A minor effect was noted on CYP2C19 activity (Ermer et al. 2015). However, we are unaware of any clinical studies directly evaluating AMP as a potential victim of a known metabolic inhibitor or as a perpetrator. For example, although AMP is at least a partial substrate of CYP2D6, there are no published clinical studies assessing the influence of a coadministered medication recognized as a CYP2D6 inhibitor (such as the serotonin-selective reuptake inhibitor [SSRI] paroxetine or fluoxetine) on the metabolism and disposition of AMP. We do know, however, from published reports that SSRIs and AMP appear to be safe and well tolerated when used concurrently (Markowitz and Patrick 2001). Additionally, although not a routine prescribing practice, the norepinephrine reuptake inhibitor atomoxetine is sometimes used in combination with a psychostimulant in patients not responding to either drug sufficiently as monotherapy. While atomoxetine-AMP combinations have not been rigorously studied or clinically evaluated for drug interaction potential, what can be said is that their combined use has been reported as generally well tolerated (Treuer et al. 2013).
Given its long history of clinical use, there are surprisingly few well-documented reports of drug–drug interactions suggestive of either metabolic inhibition or induction of AMP metabolism. Both pharmacodynamic and pharmacokinetic drug interactions with psychostimulants are extensively reviewed elsewhere (Markowitz and Patrick 2001).
Pharmacogenetic testing
Pharmacogenetic testing is increasingly affordable in the clinic and a number of testing panels directed at ADHD therapeutic agents are presently marketed. Typically, these panels include assessments of single-nucleotide polymorphisms of one or more genes related to drug metabolism (e.g., CYP2D6) and one or more genes related to drug pharmacodynamics (e.g., α-2A-adrenergic receptor gene [ADRA2A]). With respect to AMP metabolism and pharmacokinetics, there is evidence that the drug is a partial substrate of CYP2D6 with further contributions from isoenzymes of the CYP2C subfamily, FMO3, and possibly others (Fig. 1). In theory, a CYP2D6 poor metabolizer might be expected to require smaller doses of AMP and/or be more prone to adverse effects from routine clinical doses, while a CYP2D6 ultrarapid metabolizer might be anticipated to have higher dosing requirements. However, we are unaware of any published reports assessing such possibilities or documenting gene–drug concentration relationships. Accordingly, at present, the routine pharmacogenetic testing of genes encoding for drug-metabolizing enzymes involved in AMP metabolism is not recommended.
AMP excretion
AMP is eliminated through the kidneys in a pH-dependent manner. At normal urinary pH, 30%–40% of an administered dose of AMP is excreted as unchanged parent compound with hippuric acid being the second most abundant urinary metabolite. However, since AMP is a weak base with a pKa of 9.9, the urinary recovery of AMP becomes highly reliant on pH and urinary flow rates. As mentioned previously, the rate of urinary excretion of AMP is markedly influenced by urinary pH. Under conditions of acidic urine (pH <6.0), AMP is rapidly excreted, while under conditions of alkaline urine (pH >7.5), elimination of AMP is protracted (Beckett et al. 1965; Davis et al. 1971). Additionally, the relative amounts of AMP and metabolites differ based upon acid versus alkaline urine conditions.
Änggård et al. (1970) reported an increase in plasma AMP T 1/2 of ∼7 hours for every unit increase in urine pH. In addition to urinary pH, urinary volume also appeared to influence renal clearance of AMP. Such large deviations from normal physiologic urine pH seem an unlikely scenario in the absence of a purposeful effort to do so (i.e., consuming an acidifying or alkalinizing agent). However, the magnitude of the effect on AMP disposition appears far greater than any other drug–drug interaction documented for any psychostimulant (Markowitz and Patrick 2001).
Special Populations
Renal impairment
AMP is subject to both hepatic metabolism and renal elimination. LDX and d-AMP pharmacokinetics were recently reported for adults with normal kidney function and in various stages of renal dysfunction following a single 30-mg dose of LDX (Ermer et al. 2016b). Mean C max and AUC for LDX in subjects with mild, moderate, or severe renal impairment were not significantly different from those observed in subjects with normal renal function, although patients with end-stage renal disease (ESRD) had a higher mean C max and AUClast. With d-AMP, however, overall exposure (AUClast and AUC0–∞) increased and mean C max decreased as the degree of renal impairment increased. Due to the increased and prolonged exposure to d-AMP, it is recommended that downward dosing adjustments be made in individuals with impaired renal function. A further finding of this investigation was that neither LDX nor d-AMP is dialyzable (Ermer et al. 2016a).
Patient age and weight
Although there are some exceptions, the majority of pharmacokinetic studies of AMP formulations in children, adolescents, and adults have suggested that body weight is the primary determinant of differences in observed AMP plasma concentrations across the life span. In general, adults have lower overall systemic exposure to AMP when receiving the same fixed dosage and formulation of AMP, and pharmacokinetic parameters such as C max and AUC0–∞ tend to decrease with increased body weight (Kramer et al. 2005). In the case of IR AMP, available data indicate that T max values and plasma T 1/2 in children are significantly shorter (∼7 hours) than those values obtained from pharmacokinetic studies conducted in healthy adults (∼10 to 12 hours) (Brown et al. 1978, 1979; Angrist et al. 1987; Greenhill et al. 2003; Arbor Pharmaceuticals, LLC; July 1, 2016 personal communication). The elimination half-life is ∼1 hour shorter for d-AMP and 2 hours shorter for l-AMP in children relative to adults (U.S. FDA approval packet, Adderall XR, U.S. Food and Drug Administration, Center for Drug Evaluation and Research [CDER] 2001). Additionally, when children aged 6–12 years of age received a single 20-mg dose of Adderall XR, the mean C max values of d-AMP and l-AMP were 40.10 and 11.89 ng/mL, respectively (McGough et al. 2003), substantially higher than healthy adult subjects receiving the same dose in a separate study that produced values of 28.13 and 8.67 ng/mL, respectively. With the d-AMP prodrug LDX, systemic exposure was noted to be higher in younger children (6–9 years old) versus 10- to 12-year-olds. However, there seems to be relatively little difference in the metabolism and disposition of LDX in children with ADHD (Boellner et al. 2010) and healthy adults (Ermer et al. 2010) or adults with ADHD (Adler et al. 2017). In contrast, children with ADHD receiving the ODT formulation of AMP mixed isomers (AMP XR-ODT) appeared to have a far greater systemic exposure to AMP than did healthy adults receiving the same 18.8-mg dosage. Stark et al. (2017) assessed children in a single-dose, open-label, single-period pharmacokinetic study. Patients were stratified by age (6–7, 8–9, and 10–12-year-olds) and were all dosed with 18.8 mg of AMP XR-ODT under fasting conditions. The resulting C max values for d-AMP showed a decline by age group and were 102.0 ± 18.3, 89.4 ± 14.6, and 75.4 ± 18.7 ng/mL, respectively (Stark et al. 2017). Unexpectedly, an analysis indicated that patient weight was not a prominent factor in the overall decreases in AMP exposure in the three age-stratified groups (Stark et al. 2017). On the other hand, healthy adult subjects receiving the same dose of AMP XR-ODT had a mean d-AMP C max of 44.9 ± 8.9 ng/mL (Stark et al. 2016).
Sex differences in AMP pharmacokinetics
Patient sex is known to be a significant determinant in the disposition of a number of therapeutic agents (Soldin and Mattison 2009). However, significant differences between the sexes in AMP pharmacokinetics have not been rigorously investigated to date. Furthermore, the number of male subjects typically far exceeds female subjects participating in the limited number of ADHD studies, which employ pharmacokinetic sampling as part of their study design. Nevertheless, limited published studies and application materials submitted to the FDA (which include blood concentration data from both male and female subjects) have permitted a limited analysis of the issue, although in nonpeer-reviewed formats (i.e., drug-prescribing literature and CDER application materials). A general observation made with MAS in both adult healthy subjects and in pediatric patients receiving the formulation is that systemic exposure to both d- and l-AMP may be 20%–30% higher in females, but these differences are diminished when normalized by dose (mg/kg) (U.S. FDA approval packet, Adderall XR, U.S. Food and Drug Administration, Center for Drug Evaluation and Research [CDER] 2001). A similar 20%–30% higher exposure (C max, AUC) in women was reported for AMP XR-ODT with differences largely attributed to the fact that women received a higher dose on an mg/kg body weight basis (Adzenys XR-ODT 2016). With the d-AMP prodrug LDX, systemic exposure to d-AMP was about 30%–40% higher in girls than in boys. When the exposure parameters (AUC and C max) were normalized by dose (mg/kg), these differences reduced to <10%–20% (U.S. FDA approval packet, Vyvanse, U.S. Food and Drug Administration, Center for Drug Evaluation and Research [CDER] 2006; Boellner et al. 2010).
Thus, in general, it appears that females may have a 20%–40% higher exposure to a given dose of AMP; however, these differences in drug exposure tend to disappear when normalized by dose and weight (mg/kg). Nevertheless, although these differences appear to be a function of subject weight rather than a clear sex effect, the differences in exposure between sexes following the administration of a fixed dosage form are fairly significant and may be a consideration in dosage individualization.
Race and ethnicity
The influence of race and ethnicity on AMP pharmacokinetics has not been adequately studied. Limited data from both healthy adult studies and pediatric patients suggest that there are no major differences in AMP pharmacokinetics between Caucasian, black, and Hispanic populations.
Pregnancy and breastfeeding
There are limited published data on the use of AMP during pregnancy or breastfeeding. The available data are insufficient to determine if a drug-associated risk of major congenital malformations or miscarriage exists, although patients who become pregnant should avoid the use of AMP as adverse pregnancy outcomes, including premature delivery and low birth weight, have been observed in infants born to mothers dependent on AMP. Furthermore, AMP is known to readily cross the placenta and withdrawal manifestations have been reported in neonates (Jones et al. 2009; Oei et al. 2012).
It does appear that AMP is extensively transferred into breast milk (Steiner et al. 1984) and may attain concentrations that are three to seven times maternal blood concentrations. A recent case report in a 30-year-old patient treated for narcolepsy described a mother–infant pair, wherein plasma concentrations of racemic AMP were quantified in the plasma of the mother, in the breast milk, and in the plasma of the breastfed infant. The infant had plasma concentrations ∼9% that of the maternal level and no adverse effects were observed (Öhman et al. 2015). The potential for long-term neurodevelopmental effects on infants from AMP exposure is unknown and patients should be advised not to take AMP if they are contemplating becoming pregnant or do become pregnant. Additionally, individuals intending to breastfeed their infant should avoid taking any AMP formulation.
Methamphetamine
The N-methylated derivative of AMP, d-methamphetamine, received FDA approval in 1943 and presently carries an indication for the treatment of ADHD as well as exogenous obesity. Marketed as Desoxyn®, this branded name is derived from the chemical name desoxyephedrine, that is, dehydroxylated ephedrine (or diastereomeric desoxypseudoephedrine). Available as a 5-mg IR tablet only, methamphetamine is generally viewed as equipotent to d-AMP in producing behavioral stimulant effects (Kuczenski et al. 1995). However, methamphetamine is rarely used due to its questionable clinical benefit over other available AMPs and explicit safety concerns regarding its widely recognized abuse potential. Furthermore, there are no published clinical trial data documenting the clinical safety and effectiveness of methamphetamine in the treatment of ADHD (Hodgkins et al. 2012). Nevertheless, the following general pharmacokinetic attributes apply. After oral administration, methamphetamine is rapidly absorbed with a C max attained (i.e., T max) ∼3 hours following dosing and linear blood pharmacokinetics are observed. The plasma T 1/2 is similar to that of d-AMP and reported to be ∼9 to 10 hours (Cook et al. 1992; Huestis and Cone 2007). Following the oral administration of [14C] methamphetamine to two male subjects, a urinary mass balance assessment found 22% of the parent drug excreted unchanged, and the metabolite 4-hydroxymethamphetamine (15% of the dose) and minor metabolites (at 5% or less), including hippuric acid, norephedrine, 4-hydroxyamphetamine, and 4-hydroxynorephedrine (Caldwell et al. 1972). Formation of 4-hydroxymethamphetamine is believed to be catalyzed by CYP2D6 (Lin et al. 1997). In this regard, genetic polymorphisms influencing the CYP2D6 gene may be expected to influence the relative amounts of 4-hydroxy metabolites versus parent drug in the systemic circulation and urine. The clinical significance of such influences is unknown. As observed with AMP, methamphetamine excretion is highly pH dependent (Beckett et al. 1965; Kim et al. 2004).
Conclusions
AMP is one of the most thoroughly investigated molecules in all of clinical pharmacology and its pharmacokinetics has been well characterized. Presently, there are at least three different IR formulations marketed that encompass d-AMP and MAS and six MR formulations variously comprising d-AMP or mixed isomers inclusive of a prodrug, an MR oral suspension, and an ODT (Tables 1 and 2). There are a number of general observations regarding the metabolism and disposition of AMP. The drug is rapidly absorbed following oral administration and extensively metabolized into both active (minor) and inactive components with catalytic contributions from CYP2D6, isoenzymes of the CYP2C subfamily, and FMO3. There is some evidence of stereoselective metabolism of racemic AMP with d-AMP appearing to be more proficiently metabolized than l-AMP. There are no pharmacogenetic–genetic polymorphisms known to significantly influence the pharmacokinetic disposition of AMP.
In general, most studies indicate dose-proportional pharmacokinetics (i.e., C max and AUC) with most AMP dosage forms in healthy adults, adults with ADHD, and children and adolescents with ADHD. In children, adolescents, and adults, body weight appears to be the primary determinant of differences in observed AMP plasma concentrations. Although female subjects may have a 20%–40% higher systemic exposure to AMP than males on the same dose, the difference appears to be attributable to body weight. With regard to IR formulations, a typical T max is 2–3 hours and children consistently demonstrate a 60%–70% shorter plasma T 1/2 (∼7 hours) relative to adults (∼10–12 hours). Additionally, the plasma T 1/2 of the l-isomer of AMP is typically 1–2 hours shorter than that of the d-isomer. There are few pharmacokinetic drug interactions of clinical significance, with extremes in urinary pH known to produce the most significant alterations in AMP exposure (blood concentrations and elimination rate). In patients with ESRD, exposure to d-AMP is prolonged and dosing should be adjusted downward. In conclusion, the pharmacokinetics of AMP is generally dose proportional and predictable with few drug interactions or genetic polymorphisms known to significantly influence its disposition. The wide variety of AMP formulations offers the clinician significant flexibility in the pharmacotherapy of ADHD.
Clinical Significance
AMP represents one of the most well-studied and long-utilized medications in the entire psychopharmacological armamentarium. Its pharmacokinetics is characterized by rapid absorption, high absolute bioavailability, extensive hepatic metabolism, renal excretion, and dose-proportional pharmacokinetics.
There are a number of clinical take-home messages from our review. First, of the available data, it appears that female subjects typically have a 20%–40% higher systemic exposure to a given dose of AMP relative to their male counterparts. However, this difference appears to be entirely attributable to differences in body weight, which remains the best overall predictor of AMP exposure. Nevertheless, this observation should be kept in mind as it suggests the possibility that female patients may require lower overall doses of AMP to attain symptom control and, in theory, may also be more prone to side effects to the same dose of AMP received by a male patient. However, any correlation of differences in potential clinical effects with differences in drug exposure by sex remains speculative at present and requires further study.
To date, there are no known genetic polymorphisms of genes that encode for metabolic enzymes or drug transporters, which are known to significantly influence the metabolism or disposition of AMP. Additionally, there are no major pharmacokinetic drug–drug interactions recognized between AMP and other therapeutic agents.
Racemic, enantiopure, and mixed-isomer AMP salts have been formulated into a wide range of dosage formulations within the last decade, all of which have been proven safe and effective in the treatment of ADHD. There are few published head-to-head studies comparing MR-AMP formulations that incorporate both efficacy measures and pharmacokinetic measures. Accordingly, therapeutic choices are based upon the identified needs of the individual patient, including the desired period of symptom coverage. This period of coverage or symptom control is, in part, dependent on the individual formulation's pharmacokinetic release profile. The widening array of available AMP formulations permits a greater degree of drug individualization in the treatment of ADHD.
Footnotes
Disclosures
No competing financial interests exist.